انسولينوما
از تومورهاي نادر شکمي بوده که با ترشح انسولين باعث علايم شديد هيپوگليسمي و گاهي مرگ بيمار مي گردد.
محل اين تومور در غده پانکراس و با ابعاد کوچک 1 تا 2 سانتيمتر بوده و بروز ترياد ويپل علايم هيپوگليسمي، قند خون زير 50mg/dl بر طرف شدن علايم هيپوگليسمي با تجويز وريدي قند) علامت تيپيک تشخيصي آن است. اين بيماران معمولا بدليل تغذيه مواد قندي فراوان، چاق بوده و به دليل بروز اختلالات رفتاري – کاهش سطح هوشياري يا اختلالات حافظه معمولا تا مدتي با تشخيص مشکلات روحي و رواني درمان مي شوند. تشخيص هر چه سريعتر اين بيماران مهم است زيرا هيپوگليسمي مي تواند باعث آسيبهاي مغزي و حتي مرگ شود. درمان قطعي اين تومور فقط با رزکسيون آن قابل انجام است. براى مهار آزاد شدن انسولين، ديازوکسيد تجويز مىشود. براى کارسينومهاى غيرقابل درمان سلول جزيرهاي، بهترين داروى شيمىدرمانى استرپتوزوسين است.
تیروئیدیت فیبروزان مولتی فوکال :
نوعی از تیروئیدیت مزمن با مراکز فیبروز ، که بسیاری از ان ها همراه با آتیپی غیر فعال فولیکول ها می باشد.
علت آن از لحاظ پاتولوژی نامشخص است اما ممکن است تظاهری از بیماری مرتبط با IgG4 باشد .
تیروئید به علت فیبروز ، سفت و ثابت ارزیابی میشود ، ممکن است باعث ایجاد فشار به ساختمان های مجاور گردن شود.
تشخیص از طریق بیوپسی باز صورت میگیرد .
درمان آن جراحی بوده اما داروی تاموکسیفن ممکن است مفید باشد .
کارسینوم مدولاری:(MTC)
از سلولهای پارافولیکولر(سلول C)منشا میگیرد.این سلولها گیرنده TSHندارند و ید را جذب نمیکنند.اگرسلولهای بدست امده از توده تیرویید کرایتریای بدخیمی داشت و به شکل پلی گونال (چندوجهی)بوده و در سیتوپلاسم ان ماده امیلویید صورتی رنگ دیده شد تشخیص MTCاست.2نوع دارد.sporadic, familial (فامیلیال میتواند همراه با MEN2باشد.)تشخیص با FNAمی باشد.
درمان:جراحی است شامل تیروییدکتومی توتال وکمورادیاسیون .به ئلیل برداشتن تیرویید باید لووتیروکسین تجویزشود.
این نوع تومور تمایل زیادی به متاستاز به ریه،استخوان و کبد دارد.
1. فلج شدن حرکت چشم که سبب دوبینی یا تاری دید می گردد .
2. فقدان بینایی محیطی .
3. کوری ناگهانی .
4. سردرد .
5. سرگیجه .
6. فقدان هوشیاری .
اختلالات بینایی زمانی رخ می دهد که تومور بر روی عصب بینایی فشار وارد آورد . فقدان ناگهانی هوشیاری و حتی مرگ به دلیل خونریزی ناگهانی داخل تومور نیز ممکن است رخ بدهد .
فقدان هر نوع هورمون علامت خاص خود را ایجاد می کند که شامل موارد زیر خواهد بود :
1. تهوع
2. ضعف
3. کاهش یا افزایش بی دلیل وزن
4. قطع قاعدگی در خانم ها
5. ناتوانی جنسی به صورت اختلال در نعوظ
6. کاهش میل جنسی.
درمان تومورهای هیپوفیز :
جراحی : جراحی عموما از طریق استخوان اسفنوئید انجام شده و غده هیپوفیز برداشته می شود . گاهی براشتن تومور با روش کرانیوتومی انجام می شود . به این ترتیب که جراح از طریق ایجاد شکاف در جمجمه تومور را برمی دارد . این روش فقط برای تومورهای بزرگ و پیچیده انجام می شود . رادیو تراپی .
رادیوتراپی کان ون تیونال : از این روش در مواردی استفاده میشود که تومور پس از انجام جراحی مجددا عود کرده یاعلائم ناشی از آن با دارو تسکین نیابد . در این روش اشعه مستقیما به تومور تابانده می شود . این کار 5 بار در هفته و به مدت 4-6 هفته انجام می گیرد . رادیو تراپی استروتاکتیک : این یک روش جدید رادیوتراپی است که در آن از اشعه گاما استفاده می شود که مستقیما به تومور تابانده و بر روی آن متمرکز خواهد شد.
واریانت های شایع PTC:
Follicular Variant
این تومورها وقتی مورد بررسی قرار می گیرند ، مانند نئوپلاسم فولیکول هستند, از فولیکولهایی با اندازه متغیر تشکیل شده اند. کلوئید معمولاً در مقایسه با کلوئید در تیروئید غیر نئوپلاستی مجاور تیره یا hypereosinophilic است و ممکن است نمای”bubble gum” را نشان دهدسلول های غول پیکر چند هسته ای گاه به گاه در فولیکول ها وجود دارند
پیش آگهی این تومورها مشابه PTC معمولی است. یک استثناء نوع فولیکول پراکندگی یا مولتی مدولار است که دارای یک دوره بالینی تهاجمی تر است.پیش آگهی نوع فولیکولار PTC همچنین به این بستگی دارد که کاملاً محصور شده یا تهاجمی هستند
Papillary Microcarcinoma
این تومورها معمولاً به طور اتفاقی یافت می شوند و قطر آن کمتر از 1 سانتی متر است.
گاهی ممکن است بیماری همرا با متاستاز غدد لنفاوی گردن تظاهر یابد. تومورها اغلب نزدیک به کپسول تیروئید هستند. در صورت عدم محاصره بودن و وجود اسکلروز گسترده ممکن است نسبت به تومورهای کاملاً محصور شده تهاجمی تر باشد
Tall Cell Variant
این نوع توموراز سلولهایی تشکیل شده است که ارتفاعشان حداقل 2-3 برابر عرضشان است. سلولهای تومور سیتوپلاسم ائوزینوفیلی ویژگیهای هسته ای مشابه PTC معمولی دارند
Oncocytic Variant
این تومورها در معاینه شبیه به تومورهای سلول فولیکولی Hurthle هستند و از رنگ قهوه ای مشخصی برخوردار هستند. ممکن است نمای فولیکولی یا پاپیلاری داشته باشند
Columnar Cell Variant
این یک نوع نادر است که از سلولهای استوانه ای کاذب تشکیل شده است. برخی از سلولها ممکن است واکوئل های سیتوپلاسمی سوپرا نوکلئار و ساب نوکلئار داشته باشند
تیروئیدیت هاشیموتو:
این بیماری یک التهاب لنفوسیتیک اتوایمیون تیروئید است که در نتیجه ی تخریب سلول های فولیکولار تیروئید که در نهایت منجر به هایپو تیروئیدیسم میشود، رخ میدهد. تظاهر بالینی عمده ی آن بزرگ شدن منتشر و بدون درد تیروئید در زنان میانسال است. در تیروئید بیمار توده های ارتشاح لنفوسیتیک که اغلب همراه با فولیکول های حاوی مراکز زایا هستند دیده میشود، و هر دو نوع لنفوسیت های T cytotoxic و T helper افزایش پیدا میکنند. T cytotoxic باعث تخریب سلول های تیروئید میشوند و بعلاوه سلول های T به صورت موضعی سایتوکاین هایی مثل TNF ،IL1 ،INF ترشح میکنند که باعث آپوپتوز و از بین رفتن سلول های تیروئید میشود. آنتی بادی های علیه TPO و Tg در بیش از 90% موارد مثبت است که صحه ای بر اتوایمیون بودن بیماری است. در این بیماری درجات مختلفی از فیبروز مشاهده میشود.
این نئوپلاسم که با نام Hyalinized Trabecular Tumor هم شناخته می شود یک تومور نادر مشتق از سلول های فولیکولی غده تیروئید می باشد.
با توجه به ویژگی های هستهای مشابه و تغییرات ژن RET می توان آن را به نوعی یک کارسینوم پاپیلاری تیروئید محسوب کرد.(افتراق این دو از هم بسیار مشکل می باشد)
سن بروز بین ۲۱ تا ۸۰ سال متغیر بوده و زنان را به نسبت ۱.۶ به مردان مبتلا می کند. این بیماران عموماً بدون علامت هستند و شدت و نوع علائم آنها میتواند با توجه به اندازه و موقعیت آناتومیکی تومور متفاوت باشد.
بروز این آدنوم می تواند با التهاب لنفوسیتی تیروئید(تیروییدیت لنفوسیتی) و گواتر مولتی ندولر مرتبط باشد.
در اکثریت قریب به اتفاق موارد این تومور خوش خیم بوده ولی در مواردی تهاجم عروقی یا کپسولی نشان داده است.
نمای میکروسکوپی(Pathology)
●نمای ترابکولار در آن می تواند به شکل straight or curved pattern باشد
●دارای سلول های چند ضلعی با اندازه های متغیر که مرزهای سلولی در آن مشخص است.
●نمای کلیدی پاتولوژی آن سیتوپلاسم ائوزینوفیلی دارای ماده هیالن می باشد(فضای خارج سلولی هیالیننزه یا کلسیفیه شده)
●سیتوپلاسم حدود ۲ تا ۵ میکرون بوده و زرد و کروی می باشد.
●وجود شیار های هسته ای در نمای پاتولوژی بسیار متداول است.(Nulear grooves)
●ممکن است اجسام Psammona هم دیده شود.
جهت فهم بهتر slide های مربوط به نماهای پاتولوژیکی ذکر شده، آورده شده است.
■تشخیص اولیه به کمک سونوگرافی یا FNA بوده و درمان این بیماری لوبکتومی تیروئید می باشد.
پانکراس حلقوی یک بیماری نادر است که در آن قسمت دوم دوازدهه با حلقه ای از بافت لوزالمعده(عمدتا سر لورالمعده) به طور مداوم احاطه می شود. این قسمت از لوزالمعده می تواند دوازدهه را تنگ کند و جریان غذا را به سایر نقاط روده بلاک یا مختل سازد.
علائم اولیه بارز ناهنجاری شامل: پلی هیدرامانیوس (مایع اضافی مایع آمنیوتیک) ، وزن کم هنگام تولد و در مواردی همراه با استفراغ غیر صفراوی است (انسداد عموما بالای پاپیلای واتر است : بالاتر از محل اتصال مجاری صفراوی). بیماریهای مختلف کروموزومی (به عنوان مثال تریزومی 21 و با فرکانس جزئی ، تریزومی 18 و تریزومی 13) در حدود 33٪ از افراد مبتلا به پانکراس حلقوی وجود دارند.
در بزرگسالان: درد شکم در ناحیه اپیگاستریک ، حالت تهوع و استفراغ است که ممکن است قبل از رسیدن به یک تشخیص دقیق ، برای مدت طولانی (گاهی اوقات سالها) وجود داشته باشد.
این وضعیت خاص به طور معمول با رشد جنین شناسی غیرطبیعی همراه است ، اما میتواند در موارد بالغ و بعد از تولد نیز ایجاد شوند. این می تواند ناشی از :
1-رشد یک جوانه لوزالمعده شکمی در اطراف دوازدهه باشد ، جایی که قسمت هایی از جوانه شکمی با جوانه پشتی بهم متصل میشوند و یک حلقه لوزالمعده تشکیل می دهند. و یا اگر2- جوانه لوزالمعده شکمی نتواند بطور کامل چرخش کند ، بنابراین در سمت راست باقی می ماند یا اگر 3-جوانه پشتی در جهت اشتباه بچرخد ، به گونه ای که اثنی عشر توسط بافت لوزالمعده احاطه شود.
روشهای تشخیصی پس از زایمان شامل پرتونگاری و سونوگرافی شکمی ، سی تی اسکناست. رادیوگرافی شکمی می تواند نشانه کلاسیک “حباب مضاعف” را نشان دهد: وجود هوا در معده و دوازدهه. متأسفانه ، این علامت دو حبابی برای لوزالمعده حلقوی پاتولوژیک نیست ، زیرا می تواند در سایر شرایط ، مانند آترزی دوازدهه و ناهنجاری روده نیز مشاهده شود.
در نوزادان ، درمان برای تسکین انسداد معمولاً دئودنوژژونوستومی است.در بزرگسالان ، به دلیل تحرک جزئی اثنی عشر ، رویکرد گاستروژژونوستومی لاپاروسکوپی یا duodenojejunostomy است.
سودوسیست پانکراس
این عارضه بیشتر در کودکان ظاهر میشود.
علت:این عارضه معمولا بعد از پانکراتیت حاد رخ میدهد. هنگامی که توسط عفونت و یا التهاب، پانکراس تخریب شود، کیست ایجاد میشود.
بعد از آسیب به شکم و در برخی افراد با پانکراتیت مزمن، این عارضه ایجاد میشود.
علایم: نفخ شکم-
درد دائمی و یا درد شدید شکم که ممکن است به پشت هم کشیده شود-
مشکل در غذاخوردن و هضم کردن غذا
تشخیص:
احساس توده در سمت چپ و یا در وسط شکم- CT-scan -MRI – سونوگرافی
درمان: بستگی به اندازهی کیست در شکم دارد. کسیتهایی که بیش از ۶ هفته در شکم باقی میمانند و ضخامت آنها از ۵ سانتی متر بیشتر است، نیاز به جراحی دارند.
عوارض: آبسه پانکراس-ممکن است کیست پاره شود، در نتیجه بیمار دچار شوک و خونریزی بیش از حد خواهد شد.ممکن است فشار را به اندامهای دیگر منتقل سازد.
تیروئیدیت گرانولوماتوز تحت حاد ( De Quervain):
تیروئیدیت گرانولوماتوزتحت حاد به عنوان تیروئیدیت De Quervain یا تیروئیدیت سلول غول پیکر شناخته میشود .سن شایع ابتلا 30-50 سال بوده و در زنان بیشتر دیده میشود .در اکثر بیماران سابقه ابتلا به عفونت های تنفسی فوقانی وجود دارد علل ویروسی درگیر کننده شامل :ویروس Coxsackie،اوریون و آدنو ویروس ها است .
از نظرعلایم بالینی :آغاز بیماری حاد بوده و با درد گردن(هنگام بلع) ،تب ،بزرگی تیروئید ، ESR بالاو لکوسیتوز مشخص میشود .هیپرتیروئیدیسم گذرا (یعنی TSH ساپرس شده وهورمون T3,T4توتال و فری افزایش یافته ) به دنبال از هم گسیختگی فولیکول های تیروئید در برخی از افراد دیده میشود که با پیشرفت تخریب غده ممکن است به هیپوتیروئید تبدیل شود .تظاهرات بالینی در مرحله هایپرتیروئید میتواند مواردی از علایم Graves راتقلید کند.در چنین مواردی جذب و اسکن رادیونوکلئید میتواند مفید باشد زیرا تیروئیدیت تحت حاد باعث کاهش جذب ایزوتوپ و گریوزمنجر به افزایش جذب میشود .از طرفی بیماری گریوز را میتوان با رادیوتراپی درمان کرد اما تیروئیدیت تحت حاد معمولا خود محدود است و با رادیوتراپی درمان نمیشود .
از نظر پاتولوژی:گسیختگی فولیکولها و خروج کلوئید منجر به ایجاد واکنش التهابی گرانولومی و تجمع سلول های giant میشود. بارفع التهاب ،بافت فیبروز جایگزین خواهد شد.
درمان با استفاده از مسدودکننده های بتا ،آسپیرین و NSAID است (اگر جواب نداد از گلوکوکورتیکوئید استفاده میکنیم )
این وضعیت نوعا خود محدود است و در اغلب بیماران طی 6-8 به حال طبیعی باز می گردد.
پاپیلوم سینونازال (پاپیلوم اشنایدری)
این تومور خوش خیم بوده و منشا آن مخاط سینونازال است.
و میتواند بافت پوششی استوانه ای یا مطبق سنگفرشی داشته باشد.
اینگونه به نظر می آید که ویروس HPV تیپ ۶ و ۱۱ مسئول ایجاد آن میباشد.
انواع آن به ترتیب شیوع عبارتند از:
Exophytic و Inverted و سیلندری
نوع Inverted برخلاف دو نوع دیگر همانگونه که از اسمش مشخص است رشد پاپیلومایی به درون خود دارد.
علی رغم خوش خیم بودن میتواند کانونهایی از بدخیمی نیز داشته باشد و به صورت موضعی حمله به مخاط اربیت و جمجمه داشته باشد.
عود در این تومور بسیار بالاست.
اختلالات مولکولی در FTC
باز آرایی pax8/PPARy : جابه جایی کروموزومیt(2,3)[q13;p25] باعث الحاق دو ژن pax8 , PPARy می شود ژن pax8 عامل رونویسی را کد میکند که برای نمو تیروئید ضروری است و بیان ژن های اختصاصی تیروئید پروکسیداز و تیروگلوبولین را تحریک میکند ژن PPARy نیز در تمام بافت ها عامل رونویسی را کد میکند و باز آرایی این ژن در FTC نقش دارد
جهش های فعال کننده RAAS : این جهش ها هم در تومور های خوش خیم هم در بدخیم وجود دارد و به تنهایی نمیتواند باعث بدخیمی شود شاید وقوع این جهش ها یک رخداد اولیه در تومور زایی تیروئید است
جهش های پروموتر TERT: در ftc نسبت به ptc با فرکانس بالاتری وجود دارد
متیلاسیون: جهش یا حذف ژن pTEN تغییر ژنتیک کلاسیکی است که سبب فعالیت pi3K/AKT میشود و عامل تومور زایی در افراد مبتلا به این سندرم است متیلاسیون pTEN در سرطان های فولیکولار شایع است هایپر متیلاسیون RASSF1A که به عنوان سرکوبگر تومور شناخته میشود در ۷۵ درصد سرطان های فولیکولار نقش دارد
فعالیت تلومرازی از دیگر عوامل موثر در تهاجمی بودن سرطان تیروئید است افزایش فعالیت تلومراز ناشی از تغییرات بیان ژنی hTERT بررسی شده است
همچنین Mira_146b و Mir_183 ممکن است توسعه این نوع سرطان تیروئید را از طریق القای مهاجرت و مهار مرگ سلولی برنامه ریزی شده تحت تاثیر قرار دهد
سیده الهه راهنما
Riedel’s thyroiditis
تیروئیدیت ریدل, یک اختلال نادر با علت ناشناخته است که باعث فیبروز وسیع در تیرویید و نقاط دیگر بدن مثل مدیاستن و رتروپریتوئن میشود. فیبروز شدید و مهاجم در کپسول تیروئید به بافت های اطراف گسترش یافته و حتی میتواند به نای و مری و عضلات عروق گسترش یابد. علیرغم این تغییرات هیستولوژیک وسیع، اختلال عملکرد تیروئیدی شایع نیست.
از نظر بالینی خوشخیم است ولی اهمیت ان از این جهت است که غده تیروئید در این بیماری، سفت و غیرحساس (بدون تندرس)، و غالبا غیرقرینه و چسبیده و ثابت است که به دلیل چسبندگی به بافت های اطراف گردن,تقلید بدخیمی میکند و ممکن است با بدخیمی اشتباه گرفته شود.
تشخیص نیازمند بیوپسی باز است، چون بیوپسی FNA معمولا کافی نیست.
درمان این بیماری جراحی است و با هدف برطرف شدن نشانه های ناشی از فشار بر روی نای،مری و عروق و اعصاب گردن انجام میشود.(علائم فشاری :استریدور و خشونت صدا- تنگی نفس و دیسفاژی)
Undifferentiated carcinoma: تومور تمایزنیافته از منشا اپیتلیوم فولیکولی است وبسیار مهاجم است عمدتا از فقدان تومورهای دیگر در نتیجه یک یا چند تغییر ژنتیکی مثل جهش های نقطه ای غیر فعال کننده ژن سرکوبگر P53 ناشی میشوند .متوسط سن ابتلا 65 سال است .در بررسی ماکروسکوپیک توده حجیم با رشد سریع با تهاحم به کپسول و ساختارهای گردنی مشاهده میشود .در بررسی میکروسکوپیک نئوپلاسم متشکل از سلول های بسیار آناپلاستیک به صورت سلول های gaint ،سلول دوکی (نمای سارکوماتو)وترکیبی از دو نمای مزبور میباشد .
اگر بتواند ید رادیواکتیو را جذب کند میتوان از ید رادیواکتیو برای درمان سودجست و در صورتی که به رادیو تراپی با اشعه خارجی پاسخ دهد میتوان از این درمان استفاده کرد .پروگنوز بسیار ضعیف است ودر تمام موارد منجر به مرگ میشود .
تیروئیدیت حاد،یک آبسه یا سلولیت یا عفونت چرکی حاد تیروئید است.
عامل آن اغلب باکتریال است و بیمار با علائم سپسیس مراجعه میکند.
رادیاسیون ،درمان با بتا اینترفرون و آمیودارون هم از علل این بیماری هستند.
شایع ترین علت ان در اطفال و بالغین جوان ،وجود سینوس پیریفورم است، (باقی مانده ی بن بست حقی چهارم )که اغلب در سمت چپ است.
علایم بالینی شامل تب بالا،درد وتندرنس جلوی گردن تیر کشنده به گوش ،تورم یک طرفه تیروئید و دیسفاژی و اریتم روی غده است . علائم آزمایشگاهی شامل ESR بالا ،لکوسیتوز و افزایش IL_6 می باشد.
TFT نرمال (به علت عدم درگیری فولیکول ها) و اسکن تیروئید نرمال از ویژگی های تیروئیدیت حاد هستند.
تشخیص با کمک FAN ،نمونه گیری برای رنگ آمیزی و کشت صورت می گیرد .
درمان : میتوان آنتی بیوتیک تجویز کرد و اگر در CT یا سونوگرافی آبسه مشاهده شود لازم است که با جراحی آبسه را تخلیه کنیم .
پاپیلوم سینونازال ( پاپیلوم اشنایدری)
این تومور خوش خیم بوده و منشا آن مخاط سینونازال است.
و میتواند بافت پوششی استوانه یا مطبق سنگفرشی داشته باشد.
اینگونه به نظر می آید که ویروس HpV تیپ ۶ و ۱۱ مسئول ایجاد آن باشد.
انواع آن به ترتیب شیوع عبارتند از:
Exophytic و inverted و سیلندری
نوع inverted برخلاف دو نوع دیگر همانگونه که از اسمش پیداست ، رشد پاپیلومایی به درون خود دارد.
علی رغم خوش خیم بودن میتواند کانونهایی از بدخیمی داشته باشد و به صورت موضعی حمله به مخاط اربیت و جمجمه داشته باشد.
عود در این تومور بسیار بالاست.
تومور های متاستاتیک تیروئید:
اندامهای در خطر متاستاز سرطان تیروئید:
توده بدخیم تیروئید ممکن است به غدد لنفاوی، ریه ها، استخوان و گاهی مغز گسترش پیدا کند، در برخی شرایط سرطان تیروئید بسیار پیشرفته است و ممکن است به ساختار موجود در گردن مانند تراکه آ ، مری ، عروق خونی، اعصاب و یا عضلات منتقل شود، این حالت سرطان پیشرفته محل یا به اصطلاح لوکال گفته می شود.
درمان سرطان تیروئید مناستاتیک:
خوشبختانه اغلب متاستازها در ابتدا همچنان خاصیت جذب ید را دارند و به همین دلیل درمان ید رادیو اکتیو (RAI) همچنان به عنوان یک گزینه درمانی در نظر گرفته می شود، این درمان علی الخصوص برای سرطان های کوچک تیروئید با متاستاز ریه که در تصاویر اسکن نشان داده نشده اند ولی در اسکن رادیواکتیو مشخص شده است پاسخ گو خواهد بود.
متاستاز سرطان تیروئید در صورتی که پیشرفت کرده باشد و در سونوگرافی و یا CAT scan قابل مشاهده باشد، بایست تحت عمل جراحی و تیرئیدکتومی قرار بگیرد، در حقیقت جراحی روش معمول درمان متاستاز سرطان تیروئید و غدد لنفاوی در گردن است.
ریسک فاکتورها عبارتند از:
افراد بین ۲۵ تا ۶۵ سال زن بودن ، اگر به طریقی سر و گردن در معرض تابش اشعه قرار گرفته است و یا در محل بمب اتمی بودن (این عاملی است که می تواند تا ۵ سال بعد فرد را به سرطان مبتلا کند) ، داشتن گواتر و یا سابقه گواتر ، سابقه خانوادگی در بیماریها و یا سرطان تیروئید ، داشتن سوابق ژنتیکی از سرطان مانند بیماری های سرطان مدولاری تیروئید خانوادگی، سندروم نئوپلاست نوع ۲A غدد درون ریز چندگانه، سندروم نوع ۲B نئوپلاست غدد درون ریز چندگانه ،آسیایی بودن
برخی از علائم متاستاز تبروئید به اندام ها:
_متاستاز سرطان تیروئید به ریه:
سرفه به شکلی که قطع نشود ،تنگی نفس، عفونت مکرر قفسه سینه،سرفه های خونی، درد یا ناراحتی در قفسه سینه،کاهش وزن
_متاستاز سرطان تیروئید به استخوان :
درد استخوان،شکنندگی استخوان،مشکلات عصبی
ادنوم فولیکولار تیروئید:
ادنوم فولیکولار تیروئید یک تومور خوش خیم بدون کپسول غده ی تیروئید است و یکی از نئوپلاسم های شایع تیروئید میباشد و به دو گروه عملکردی و غیر عملکردی تقسیم بندی میشود که عملکردی همان ادنوم فولیکولار توکسیک است و وباعث هایپر تیروئیدی علامت دار میشود و 1%فولیکولار ادنوما هارا دربرمیگیرد.اتیولوژی:کمبود ید و گواتر اندمیک فاکتور های زمینه ساز کنسر های فولیکولار است و فعالیت انکوژن ها و موتاسیون های ژنی در ادنوم فولیکولار شایع است.علائم:یک ندول سفت و قابل لمس تیروئید در معاینه میباشد و عموما بدون علامت اند مگر اینکه توده بزرگتر باشد و علائمی مانند دیس پنه و خشونت صدا و درد گردن و دیس فاژی بروز کند.تشخیص با استفاده از اسپیراسیون سوزنی تیروئید و سونوگرافی و اندازه گیری سطح TSH سرم میباشد.درمان :شامل لوبکتومی تیروئید و Isthmusectomy بوده و در بیماران ادنوم فولیکولار عملکردی میتوان ید رادیواکتیو 131 تجویز کرد و نیاز به لوبکتومی نیست.در سیر بیماری ادنوم فولیکولار غیر عملکردی ممکن است به کارسینوما تبدیل شود.
PTCشایعترین نوع سرطان تیروئید است. با اینکه رشد آن کند است ولی باز هم در صورت عدم درمان امکان پیشرفت دارد
این سرطان از سلول های تیروئید آغاز و ممکن است بجز تیروئید به غدد لنفاوی یا ریه کبد و مغز استخوان نیز سرایت کند
علایم: در اکثر موارد بدون علامت است ولی معمول ترین علامت آن وجود توده در گردن به صورت توده جامد نامنظم و یا چیتیک است که از رشد غیر طبیعی بافت به وجود می آید
سن رایج این سرطان بین ۳۰ تا ۵۰ سال است و در زنان شایع تر است درمان بستگی به زمان تشخیص دارد اگر توده از یک و نیم سانتی متر کمتر باشد درمان با پیش آگهی خوب امکان پذیر است و زیر ۵۵ سال به درمان بهتر پاسخ میدهند
درمان اولویت دار جراحی تیروئید است
اگر توده کوچک تر از یک سانتیمتر باشد یا به هر دلیلی امکان جراحی نباشد یا با وجود جراحی دچار شده باشد می توان از درمان kf تیروئید یا مایکروویو استفاده کرد
تشخیص :FNA سونوگرافی داپلر،mri،ct تیروئید برای بررسی میزان هورمون ها و فاکتورهای خون
بیماری پانکراتیت حاد
فعال سازی نامتناسب پیش ساز های آنزیم های پانکراسی , درون پانکراس باعث خود هضمی ,نکروز , ادم و خونریزی می شود.
شایع ترین علت آن مصرف زیاد الکل یا سنگه های صفراوی و بیماری های صفراوی می باشد .
نشانه های پانکراتیت حاد شامل ۱_ درد شدید و ثابت اپیگاستریک که یک الی چهار ساعت پس از صرف غذا یا مصرف الکل زیاد آغاز می شود ۲- کاهش درد به هنگام خم شدن به جلو ۳ – تهوع و استفراغ ۴-انتشار درد به پشت ۵ -کبودی پهلوها و اطراف ناف…
تشخیص پانکراتیت حاد :
۱.ام آر آی و سی تی مثبت به همراه آمیلاز سرمی بالا تسخیص دهنده است .
۲.آمیلاز بالای ۱۰۰۰ نشان دهنده ی بیماری های صفراوی می باشد.
۳.نسبت آمیلاز به کراتینین ممکن است حساسیت تشخیص را افزایش دهد .
۴.افزایش LFT
۵.انجام ERCP
درمان پانکراتیت حاد:
۱.انجام NPO
۲. مایع درمانی
۳.آنتی بیوتیک ها
۴.ساکشن نازوگاستریک
۵.درمان هایپر گلایسمی و هایپو کلسمی همراه.
پیش آگهی و سیر بالینی:
موارد خفیف در مدت یک هفته برطرف می شوند .
موارد شدید ممکن است باعث شوک و سپسیس یا حتی مرگ شوند.
معیار های رانسون برای ارزیابی شدت بیماری:
اگر کمتر از ۳ معیار وجود داشته باشد میرایی کم تر از ۱ درصد و اگر معیار ها بیش از ۶ عدد باشد میرایی تا حد ۱۰۰ درصد افزایش می یابد..
ممکن است کیست و آبسه پانکراس ۳ تا ۶ هفته بعد ایجاد شود .
ممکن است که پانکراتیت حاد به فرم مزمن در آید.
پانکراس دوقسمتیPancreas divisum)):
تعریف و اپیدمیولوژی:
شایع ترین تغییر شکل آناتومیک مادر زادی پانکراس انسان می باشد که در آن در دوران جنینی نیمه های شکمی و پشتی پانکراس بهم نمی چسبند. بدین ترتیب درناژ پانکراس از طریق پاپیلا های فرعی صورت می گیرد و در واقع در این ناهنجاری یک مجرای منفرد پانکراسی نداریم و اغلب در عوض آن دو مجرای مجزای پشتی و شکمی داریم. این اختلال در 7-10 درصد از جمعیت دیده می شود.
علایم بالینی و تشخیص:
در اکثر موارد ، بیماران بدون علامت هستند اما در بعضی موارد علایمی مانند درد شکم ، تهوع و استفراغ بروز می یابد . این آنومالی در اکثر بیماران آن هارا مستعد ابتلا به پانکراتیت نمی کند، اما ترکیبی از پانکراس دو شاخه و سوراخ فرعی کوچک می تواند باعث انسداد مجرای پشتی شود و فرد را مستعد ابتلا به پانکراتیت حاد و مزمن کند.
تشخیص با مشاهده وجود دو مجرای فرعی مجزا در پانکراس با استفاده از تکنیک های MRCP Magnetic Resonance) Cholangiopancreatography) و یا ERCP((Endoscopic Retrograde Cholangiopancreatography مسجل می شود .
درمان:
اگر با علایم بالینی همراه نباشد نیاز به مداخله ای نیست . اما در صورت بروز علایم بالینی مداخله اندوسکوپیک یا جراحی اندیکاسیون پیدا می کند.
سومین نوع توموری که در پانکراس ایجاد می شود گلوکاگونوما (به انگلیسی: Glucagonoma) یک تومور نادر سلولهای آلفای لوزالمعده است که منجر به تولید بیش از حد هورمون گلوکاگون میشود. تومورهای سلولهای آلفا معمولاً همراه با سندرم گلوکاگونوما دیده میشوند
علائم و نشانهها
تولید بیش از حد هورمون گلوکاگون منجر به افزایش قند خون از طریق فعال شدن فرایندهای آنابولیک و کاتابولیک بدن، از جمله گلوکونئوژنز و تجزیه و تحلیل چربی میشود. گلوکونئوژنز تولید گلوکز از پروتئین و اسیدهای آمینه میباشد. تجزیه و تحلیل چربیها نیز افزایش مییابد. نتیجه این میشود که سطح بالایی از گلوکاگون، کاهش سطح خونی اسیدهای آمینه و کاهش تعداد گلبولهای قرمز خون همراه با اسهال و کاهش وزن تا ۱۵ کیلوگرم را در بیمار میشود مشاهده کرد.
راش (لکه پوستی) در نواحی مختلف بدن، زخم زبان
علت
اگر چه علت این بیماری ناشناخته است، عوامل ژنتیکی نقش مهمی در برخی از موارد بازی میکند. سابقه خانوادگی نئوپلازی متعدد نوع یک غدد درون ریز میتواند عامل خطر باشد. این تومورها معمولاً بدخیم هستند.
تشخیص
سرم خونی گلوکاگون با غلظت ۱۰۰۰ pg./میلی لیتر یا بیشتر نشان دهنده گلوکاگونوما میباشد. (محدوده نرمال بین ۵۰ تا 200 pg/mL میباشد.
در CBC میتوان علائم کم خونی را مشاهده کرد.
لکه پوستی در تشخیص گلوکوناما از اهمیت فراوانی برخوردار است.
موقعیت تومور را میتوان به وسیله ابزارهای رادیوگرافی از جمله آنژیوگرافی، سی تی اسکن، ام آر آی یا پت اسکن مشخص نمود.
درمان
تنها راه درمان برای گلوکاگونوما جراحی و برداشتن کامل تومور ست.
Pituicytoma توموري نادر در لوب خلفی هيپوفيز است که از سلولهای بزرگ كف الود تشکیل شده است و همچنين به ان infundibuloma مي گوييم. این موارد به ندرت در طول زندگی بيمار مشخص می شوند و اغلب اوقات در کالبد شکافی و MRI كه به علت هاي ديگر انجام مي شود كشف مي شوند.این تومورها ناشی از سلول هاي pituicytes هستند که سلولهای استرومائی (پشتیبان) لوب خلفی هستند. آنها ممکن است با نقص میدان بینایی ، سردرد ، كم كاري هيپوفيز و یا در طول زندگی بدون علامت باشند آنها معمولاً برخلاف موقعیت مکانی اي که دارند باعث ديابت بي مزه نمي شوند. به نظر می رسد که جراحی روش درمانی مطلوب است اما از آنجا که بسیار نادر هستند ، درمان قطعي براي ان ها و جود ندارد. آنها نسبت به آدنوم هاي استاندارد هیپوفیز كه ناشی از لوب قدامی غده هستند تمايل عروقی بيشتري دارند و به اين دليل جراحی ان ها سخت تر است.
Pituicytoma توموري نادر در لوب خلفی هيپوفيز است که از سلولهای بزرگ كف الود تشکیل شده است و همچنين به ان infundibuloma مي گوييم. این موارد به ندرت در طول زندگی بيمار مشخص می شوند و اغلب اوقات در کالبد شکافی و MRI كه به علت هاي ديگر انجام مي شود كشف مي شوند.این تومورها ناشی از سلول هاي pituicytes هستند که سلولهای استرومائی (پشتیبان) لوب خلفی هستند. آنها ممکن است با نقص میدان بینایی ، سردرد ، كم كاري هيپوفيز و یا در طول زندگی بدون علامت باشند آنها معمولاً برخلاف موقعیت مکانی اي که دارند باعث ديابت بي مزه نمي شوند. به نظر می رسد که جراحی روش درمانی مطلوب است اما از آنجا که بسیار نادر هستند ، درمان قطعي براي ان ها و جود ندارد. آنها نسبت به آدنوم هاي استاندارد هیپوفیز كه ناشی از لوب قدامی غده هستند تمايل عروقی بيشتري دارند و به اين دليل جراحی ان ها سخت تر است
clear cell tumor
clear cellدر پاتولوژی به منظور دیدن سلول هایی است که سیتوپلاسم آنها در رنگ آمیزی H&E به صورت شفاف دیده میشوند.clear cell ها در واقع سلول هایی با قابلیت ترشحی در اپیتلیوم هستند و یکی از اجزای غدد عرقی نیز محسوب میشوند.
احتمال بروز این درگیری به طور شایع در 70 – 80 درصد کارسینوماهای کلیه و همچنین در تخمدان و بافت نرم و پوست نیز وجود دارد.
در کلیه به صورت افزایش ناگهانی در تعداد سلول ها تظاهر میابد که منجر به تشکیل mass(توده) میشود.دلیل اصلی آن ناشناخته میباشد اما عواملی از قبیل سیگار ، بعضی داروها و بیماری های ارثی مانند ون هیپل لیندو موثر اند.
renal clear cell carcinoma میتواند به صورت فامیلیال(5%) و اسپورادیک(95%) رخ دهد که به دلیل اختلال در بازوی کوتاه کروموزوم 3 رخ میدهد.در صورتی که به علت ون هیپل لیندو (VHL) باشد اختلال در 3p25 از کروموزوم 3 میباشد.
برای درمان در خط اول جراحی را خواهیم داشت در صورتی که نتوان به طور کامل توده را خارج کنیم برای FOLLOW UP از مواردی چون رادیوتراپی و کموتراپی استفاده میکنیم
متاستاز سرطان تیروئید به غددلنفاوی
گسترش سرطان تیروئید به غدد لنفاوی می تواند به دو شکل نمود داشته باشد، در حالت ماکروسکوپی غدد لنفاوی دچار تورم شده و بزرگتر از حد معمول هستند و تورم دیده می شود، در حالت میکروسکوپیک لنف نود ها در سایز تغییری نمی کنند و تنها در بررسی های میکروسکوپیک مشخص می شوند، نرخ عود مجدد بیماری در دو حالت متفاوت است.
البته وجود یک توده در گردن نشانه سرطان نیست بلکه یک هشدار برای بررسی بیشتر است ولیکن علائمی نیز ممکن است بیمار را درگیر نماید این علائم عبارتند از تب، لرز، تعیق شبانه، خستگی و کاهش وزن، همچنین وجود گره های لنفاوی بزرگ در بدن
درمان
با وجود عوارض زیاد هنوز هم خط اول در درمان سرطان تیروئید جراحی است ولیکن، به تازگی پزشکان به روشهای کمتر تهاجمی مانند آر افو ماکروویو پی برده اند .
از روش آر اف در موارد زیر استفاده می شود:
• به علت عود مکرر چندین مرتبه جراحی شده و به دلیل چسبندگی و فیبروز امکان عمل جراحی وجود ندارد
• غدد در نواحی هستند که دسترسی جراح به آنها بسیار مشکل است مانند زون ۶
• و یا اینکه غدد لنفاوی بدخیم زیر ۱۰ میلی متر هستند و ممکن است جراح آن ها را در حین جراحی پیدا نکند، این موارد از جکله مواردی هستند که برای آنها درمان با آر اف در نظر گرفته می شود.
تیروئیدیت لمسی یا palpation thyroiditis
واژه palpation به معنای تخمین سختی بافت بیمار با استفاده از دستان متخصص است.
گسترش التهاب در تیروئید در اثر اسیب مکانیکی به فولیکول های تیروئید میباشد.این اتفاق ممکن است در اثر پالپیشن مکرر و شدید تیروئید یا مداخله جراحی مثل دایسکشن رادیکال گردن بیوفتد.(یعنی خارج کردن گره های لنفاوی جهت بررسی متاستاز)
نوعی تیروئیدیت تحت حاد است .*در تیروئیدیت تحت حاد، در غده تیروئید یک ارتشاح یا اینفیلتراسیون التهابی مشخص تکه ای در نقاط مختلف دیده می شود که با تخریب فولیکول های تیروئید و گلبول های سفید بزرگ چند هسته ای در داخل بعضی از فولیکول ها همراه است. تغییرات فولیکولی به سمت تشکیل گرانولوم پیش رفته و در نهایت منجر به فیبروز می شوند.
علائم تیروئیدیت تحت حاد: در تیروئیدیت تحت حاد، بیمار معمولا با یک تیروئید بزرگ و دردناک مراجعه می کند و از درد گردن شاکی است. گاهی تب نیز دیده می شود.تشخیص بیماری بر اساس موارد زیر صورت میگیرد:علائم و نشانه های بیماری: دردگلو با انتشار به گوش و فک، حساسیت در لمس، گواتر، علائم تیروتوکسیکوز یا کم کاری تیروئید.
آزمایش ها: ESR بالا و لکوسیتوز و معمولا منفی بودن Anti TPO و آزمایش تیروئید (T3 ، T4 ، TSH).
در نهایت در صورت شک به تشخیص می توان از FNA و جذب ید رادیواکتیو (اسکن تیروئید) استفاده کرد. در FNA تیروئیدیت تحت حاد، سلول های بزرگ چند هسته ای دیده می شود و در اسکن تیروئید معمولا جذب ید رادیواکتیو کاهش یافته است.
*پاتولوژی تیروئیدیت لمسی multifocal granulomatous folliculitisرا نشان میدهد. سلولهای T در مقایسه با سلولهای B غالب هستند. ممکن است پرکاری تیروئید زودگذر به دلیل نشت هورمون پیش ساخته تیروئید در خون وجود داشته باشد.
FMTC بیماری ارثی آتوزومی غالب می باشد. بر اثر جهش در پروتوانکوژن RET ایجاد میشود.
کارسینوم مدولاری تیروئید در 70% موارد تک گیر و در 30% باقی مانده یه صورت خانوادگی و در زمینه ی سندرم نئوپلاسم های متعدد اندوکرین (MEN) 2A یا 2B یا کارسینوم مدولاری تیروئید خانوادگی (FMTC) بدون همراهی با سندرم MEN روی می دهد. هر دو شکل خانوادگی و تگ گیر دارای جهش های فعال کننده ی RET می باشند. کارسیونوم مدولاری تک گیر و موارد خانودگی بدون ارتباط با سندرم MEN در بالغین دیده می شود و حداکثر میزان بروز آن دهه های پنج و شش عمر می باشد. برعکس، مواردی که همراه MEN-2A و MEN-2B هستند در سنین پایین تر و حتی کودکان دیده می شوند.
تومور MTC نئوپلاسم هایی نورواندوکرین هستند که از سلولهای C یا پارافولیکولار تیروئید منشاء میگیرد. سلولهای کارسینوم مدولاری تیروئید، اغلب قوام سفت و بدون کپسول دارند و هورمون کلسیتونین ترشح میکنند.
ریخت شناسی :
کارسینوم های مدولاری ممکن است به صورت ندول های منفرد یا ضایعات متعدد باشند که هر دو لوب تیروئید را گرفتار کرده اند.
** نمای مشخصه ی این تومور ها رسوبات آمیلوئید که از مولکول های کلسیتونین تغییر شکل یافته تشکیل شده اند می باشد.
** ویژگی خاص کارسینوم مدولاری خانوادگی حضور هیپرپلازی چند کانونی سلول C در پارانشیم تیروئی مجاور است. این ویژگی معمولا در تک گیر دیده نمی شود. به نظر می رسد که کانون های هیپرپلازی سلول C به عنوان ضایعات پیش ساز کارسینوم مدولاری باشند. تشخیص پاتولوژی با رنگ آمیزی ایمونوهیستوشیمیایی کلسیتوئین و مشاهده آن درون سیتوپلاسم سلول های تومور و در آمیلوئید استرومایی انجام می شود.
تظاهرات بالینی:
• ندول سفت یا توده در تیروئید.
• بزرگی غدد لنفاوی گردن.
• اغلب توده های گردنی دردناک هستند.
• گاهی 2 طرف تیروئید درگیر میشود.
• اغلب در 3/2 فوقانی غده تیروئید دیده شوند.
گواتر دیس هورمونوژنتیک
بزرگ شدن تیروئید به دلیل نقص های مختلف ارثی در هورمون تیروئید است که عدم گردش هورمون تیروئید موجب ترشح tsh میشود که این امر موجب تخریب بیش از حد و هایپرپلازی تیروئید میشود و علت نقص در ژن های رمز کننده انزیم هایی است که در سنتز هورمون های تیروئیدی نقش دارند بیشتر در کودکان است . در دوران نوزادی با گواتر مشهود و کم کاری مادرزادی تیروئید اختلال در رشد و ناتوانی ذهنی همراه است در بزرگسالی گواتر و شواهد کمتری از اختلال عملکرد تیروئید وجود دارد
تشخیص: سابقه خانوادگی- غربالگری های روتین نوزادان در رارزیابی رادیولوژیک شامل سیتی و سونو و اندازه گیری tsh درمایع امنیوتیک در رحم میتوان تشخیص داد
درمان: جایگزین t4تا زمانی که tshنرمال شود -بعضی با ید درمان میشوند- در انواع بزرگ جراحی- و در گواتر جنینی تزریق تیروکسین ب مایع امنیوتیک
يكي از انواع كارسينوماي فوليكولار تيروئيد است،سلول هاي اكسيفيليك در تيروئيد اغلب هرتل سل ناميده ميشوند.در اين تومور سلول هاي اكسي فيليك دچار افزايش سايز با گرانول هاي سيتوپلاسمي ائوزينوفيلي مشخص ميشوند.
اغلب اين تومور با تيروئيديت هاشيموتو همراه است .
انواع مختلفي از خوش خيم تا بدخيم ميتواند داشته باشد،اما معمولا خوش خيم است و بعد جراحي عود نميكند.
علت مشخصي ندارد اما ميتوا رادياسيون سر يا گردن ،كمبود يد،موتاسيون ژنتيكي يا سابقه سرطان فاميلي را به عنوان محرك در نظر گرفت.
بيشتر درسنين بالاو ٧٠ تا ٨٠ سال ديده ميشود.
پانکراس هتروتروفیک یک بیماری نسبتاً نادر است . میزان بروز آن در مجموعه کالبد شکافی 0.5-13.7٪ است.
شایع ترین محل آن معده (38/25 درصد) و بعد آن دوازدهه (36/17 درصد) ، ژژنوم (15.21.7 درصد) و بندرت در مری ، مجرای صفراوی مشترک ، طحال ، مزنتری ، مدیاستن و لوله فالوپ است. در مورد پاتوژنز هتروتوپی پانکراس نظریه های مختلفی وجود دارد. دو فرضیه پیشنهادی محتمل که در مورد بافت پانکراس هتروتروفیک توضیح می دهند عبارتند از: 1) جوانه های بافت پانکراس جنینی جدا شده در حین چرخش foregut و همجوشی قسمت پشتی و شکمی لوزالمعده متعاقباً به طور مستقل به عنوان جزایر بافت پانکراس در دستگاه GI رشد می کند. 2) یک استعمال نامناسب لوزالمعده از بافت آندودرمال هنگام جنین زایی
نوع اول از بافت پانکراس معمولی با سلولهای acini ، مجاری و جزایر تشکیل شده است. نوع II مجرای لوزالمعده را نشان می دهد. نوع III که فقط از بافت آکینار تشکیل شده است. نوع IV تنها جزایر (پانکراس غدد درون ریز) است.
لوزالمعده هتروتروفیک معمولاً بدون علامت است. درد شایعترین علامت است و علائم دیگر ممکن است به دلیل اثر انسداد مانند انسداد روده معده و زردی انسدادی ایجاد شود. عوارض نادر شامل خونریزی ، پانکراتیت ، تصورات درونی ، انسولینوما ، سندرم زولینجر-الیسون ، کیست شبه پانکراس آبسه لوزالمعده و تحول بدخیم در بافت لوزالمعده هتروتروفیک است.
تشخيص افتراقي شامل تومور استرومايي دستگاه گوارش (GIST) ، تومور عصب اتونوم دستگاه گوارش (GANT) ، کارسينوئيد ، لنفوم و آدنوکارسينوما است [7]. در آندوسکوپی ، یک ضایعه زیر مخاطی عمود بر پایه ذکر شده است. سونوگرافی آندوسکوپی (EUS) یک عامل تشخیصی مفید در شناسایی ضایعه محسوب می شود. EUS و CT اسکن همزمان یک روش برتر است. بیوپسی معمول آندوسکوپی ممکن است سطحی باشد و منجر به نتیجه نادرست شود. با این حال ، بیوپسی جامبو در بسیاری موارد می تواند کمک کننده باشد. بیوپسی منجمد از عمل جراحی می تواند این تشخیص را ایجاد کند. ضایعات علامتی و مشکوک برای برداشتن جراحی در نظر گرفته می شوند. .
بیماری Graves یک اختلال سیستم ایمنی است که سبب تولید بیش از حد هورمون هلی نیرویی (هایپرتیروییدیسم) می شود. گرچه اختلالات مختلفی می تواند به هایپرتیروئیدیسم منجر شوند، بیماری Graves یک علت شایع آن است.
این بیماری هرفرد را می تواند مبتلا کند اما در خانم ها و در سن زیر ۴۰ سال شایع تر است.
مکانیسم این بیماری افزایش آنتی بادی محرک گیرنده های تیروئید(TSI) است که مشابه TSH(هورمون محرک تیروئید) عمل می کند.
دو مشخصه بارز آن افتالموپاتی و درماتوپاتی است که با مشاهده آن ها می توانیم به تشخیص برسیم. علایم بالینی آن عبارت اند از: بی قراری و عدم تحمل به گرما، ترمور، تاکی کاردی، کاهش وزن، بزرگ شدن غده تیروئید (گواتر)، اگزوفتالمی و… .
در این بیماری سطح هورمون T3 و T4 افزایش و هورمون محرک تیروئید (TSH) کاهش پیدا می کند.
برای درمان از داروهایی مانند متیل مازول، پروپیل تیواوراسیل و کاربیمازول استفاده می شود.
Pituitary hyperplasia هیپرپلازی هیپوفیز : افزایش غیر نئوپلاستیک در تعداد سلول های آدنوهیپوفیز( هیپوفیز قدامی )
ویژگی های ضروری: هیپرپلازی ندولار بدون فشار آوردن به بقیه نواحی یا یک افزایش منتشر سلول های هیپوفیزی بدون از بین رفتن ساختار غده
اپیدمیولوژی: به طور معمول بسیار نادر در نظر گرفته می شوند اما تحقیقات کالبد شکافی شیوع را به میزان 26% گزارش می دهد و ممکت است در هایپرپلازی پرولاکتین در بارداری شایع تر باشد. به طور معمول در بالغین جوان مشاهده می شود.
محل : آدنوهیپوفیز
پاتوفیزیولوژی:
پاسخ فیزیولوژیک به یک از کار افتادگی عضو انتهایی:
*شایع ترین » هیپرپلازی پرولاکتین در بارداری ، شیردهی یا استروژن درمانی
*هیپرپلازی سلول های ACTH به دلیل کمبود کورتیزول در بیماری آدیسون
*هیپرپلازی سلول های TSH همراه با هیپوتروئیدیسم اولیه طولانی مدت
*هایپرپلازی گنادوتروپ در هیپوگنادیسم اولیه به دلیل سندروم های کلاین فلتر و ترنر
هایپرپلازی پاتولوژیک همراه با هورمون های آزادکننده زیاد ترشح شده از جایی غیر از هیپوتالاموس:
*هایپر پلازی سلول های هورمون رشد به دلیل افزایش هورمون آزاد کننده هورمون رشد ترشح شده توسط تومور سلول های جزایر پانکراس ، فئوسیتوکروما، تومور های کارسینوئید و برونشیال
*هایپرپللازی سلول های ACTH به دلیل ترشح هورمون آزادکننده کورتیکوتروپین از هامارتوم هیپوتالاموس یا تومور های نورواندوکرین
ایدیوپاتیک
ارثی
ویژگی های بالینی:
هایپرپلازی های مرتبط با هورمون
سلول های هورمون رشد: غول آسایی یا آکرومگالی
سلول های پرولاکتین: hyperprolactinemia هایپر پرولاکتینمی
سلول های ACTH : سندرم کوشینگ
سلول های TSH : hyperprolactinemia
کم کاری تیروئید اولیه طولانی مدت منجر به هایپرپلازی TSH می شود
عدم وجود بازخورد منفی از تیروکسین (T4) منجر به افزایش سطح TRH می شود که سلولهای TSH و PRL هیپوفیز را تحریک می کند.
LH-FSH : نتایج اولیه شروع هیپوگنادیسم
تشخیص
بیوپسی
به ندرت با جراحی خارج می شود
به احتمال بسیار زیاد در کالبد شکافی شناسایی می شود
درمان :
معمولا اختلال زمینه ای اندوکرینولوژیک را باید تصحیح کرد
جراحی : برای هایپرپلازی سلول های هورمون رشد و ACTH
درمان کمکی : دارو درمانی برای کم کاری تیروئید
آدنوم هیپوفیزی (pituitory adenoma)
آدنوم های هیپوفیزی توموهای نورواندوکرین هیپوفیز قدامی هستند که از سلول های هیپوفیزی ترشح کننده هورمون تشکیل شده اند. تیپ های اصلی این تومورها شامل سوماتوتروپ، لاکتوتروپ، تیروتروپ، کورتیکوتروپ، گنادوتروپ، سلول های خنثی (Null) و چند هورمون (plurihormonal) هستند. آدنوم های هیپوفیزی 10 تا 15% نئوپلاسم های داخل جمجه را تشکیل می دهند. شیوع آن ها در دهه ی چهارم تا هفتم زندگی است. میزان شیوع اندکی در زنان بیشتر است. اکثر این تومورها در زین ترکی و درون لوب قدامی هیپوفیز یافت می شوند، گرچه موارد نادر در ساقه هیپوفیزی و حتی به عنوان بخشی از تراتوماهای تخمدانی نیز گزارش شده اند. اکثر بیماران (میکروآدنوماها) تظاهرات افزایش هورمون را نشان می دهند. تومورهای بزرگ تر (ماکروآدنوماها، اندازه بیش از 1 سانتی متر) اثراتی همچون سردرد یا اختلال در بینایی نیز ایجاد می کنند. نکروز هموارژیک تورموهای بزرگ یک اورژانس جراحی تلقی می شود. در بررسی های آزمایشگاهی اندازه گیری سطح پرولاکتین سرم، ACTH سرم، هورمون های تیروئیدی و… برای تشخیص استفاده می شود. در بررسی های میکروسکوپی باید یافته های بافت شناسی معمول مثل درجه میتوز، سلول های ژانت، انکولوزیون ها، خونریزی و ویژگی های عروقی مد نظر قرار گیرد. یکی از راه های درمان این تومورها جراحی است.
۵ سلول موجوددرهیپوفیز قدامی:
1 .سوماتوتروپ ها 30 تا 40 درصد
2 .کورتیکوتروپ ها 20 درصد
3 .تایر توپها 3 درصد
4 .گنادوتروپ و لاکتوتروپ ها
یکی از تظاهرات بالینی توده های هیپوفیز،Hemianopsia Hemolimusاست که بخاطر اثرات فشاری توده ایجاد میگردد. در ناحیه تمپورال نیز ممکن است آسیب ایجاد کند و فرد شنوایی خود را ازدست بدهد. که اثرات بستگی به سایز توده دارد.
توده هایی که درهیپوفیزترشح دارند:
.1آدنوم هایی که ترشح کننده هورمون هستند
ممکن است پرولاکتین ترشح کنند که منجر به آمنوره، گاالکتوره می شود.
هورمون رشد:اگر قبل از بلوغ باشدمنجر به ژیگانتیسم
اگر بعداز بلوغ باشد تبدیل به آکرومگالی می شود.
کورتیزول ایجاد کوشینگ و سندروم nelsonمی کند
TSH هایپرتیروئیدیسم را سبب می شود.
2 .ممکن است آدنوم هایی داشته باشیم که هیچ گونه ترشح هورمونی ندارد که 20 تا 25 درصد بیماران از این دسته هستند
معمولا علامتی برای بیماران ندارند و زمانی به تشخیص میرسیم که سایز توده بزرگ شود و مشکالت زیادی برای بیمار ایجاد کند زمانی علامت دار می شود که اثرات فشاری دارند یعنی زیاد بزرگ می شود و منجر به اختلالات بینایی سردرد و کمبود های هورمونی می گردد.
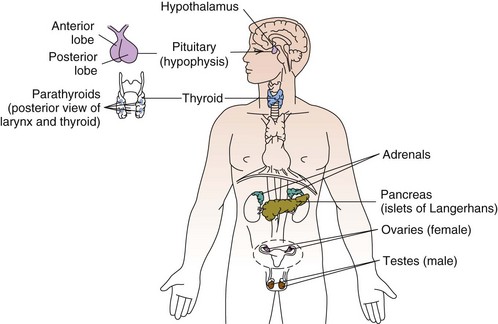
ناحیه ی پشتی بن بست سوم به غده ی پاراتیروئید تحتانی تمایز می یابد. ناحیه ی شکمی بن بست سوم هم تیموس را می سازد.
بخش پشتی بن بست چهارم غدد پاراتیروئید فوقانی را می سازد. بخش شکمی بن بست چهارم جسم اولتیموبرونشیال را می سازد که بعدا به سلول های c تیروئید که مسئول ساخت کلسی تونین است، تبدیل می شوند.
آنومالی های تیموس که منجر به تیموس گردنی نابجا می شود ، در اثر سه مکانیسم زیر ممکن است به وجود آید:
1)نزول ناکامل تیموس به قفسه ی سینه
2) باقی ماندن قسمتی از بافت تیموس در مسیر طبیعی نزول تیموس پبه قفسه سینه
3)از بین نرفتن مجرای تیموفارنژیال
آنومالی بعدی کیست های تیموسی است که 2 فرضیه جهت توجیه پاتوژنز آنها وجود دارد:
1)تخریب پیشرونده و اکتسابی باقی مانده های اپیتلیالی یا اجسام هاسال(hassall)
2)بقایای مجرای تیموفارنژیال تشکیل کیست می دهد.
آنومالی پاراتیروئید به صوت ایجاد کیست های پاراتیروئیدی تظاهر میکند.
از لحاظ جنین شناسی این کیست ها یا غدد پاراتیروئید نا بجا می توانند در هر جایی از اطراف عده ی تیروئید پیدا شوند ولی رایج ترین مکان در بخش پایینی غده ی تیروئید است.
به خاطر ارتباط خاستگاهی پاراتیروئید و تیموس، در هرجایی که آنومالی تیموس داشته باشیم امکان وجود آنوملی پاراتیروئید نیز وجود دارد.برای مثال می تواند در مدیاستن تیموس و پاراتیروئید نا بجا پیدا کرد.
کیست های پارا تیروئیدی معمولا از نظر بیوشیمیایی مشکلی ندارند ولی گزارشاتی مبنی بر پاراتیروئیدی ناشی از این کیست ها وجود دارد.
پاتوژنز این کیست ها چند فرضیه را شامل می شود:
1)بقایای جنینی بن بست حلقی سوم و چهارم
2)تخریب آدنومای پاراتیروئید به صورت کیستیک
3)تجمع ترشحات غده وایجاد میکروکیست. به مرور این میکروکیست ها بزرگ شده و کیست را ایجاد می کنند. یا اینکه چند میکروکیست به هم متصل شده و کیست بزرگی را به وجود می آورند.
Scc تیروئید
بسیار نادر ، بسیار کشنده و تماما از سلولهای سنگفرشی تشکیل شده است. از نظر بالینی و آسیبشناسی اشتراکاتی با سرطان آناپلاستیک تیروئید دارد و میتوان آن را به عنوان نوعی از سرطان آناپلاستیک تیروئید در نظر گرفت . مواردی که باید rule out شود :
• متاستاز یا تهاجم مستقیم از سلولهای سنگفرشی اوروفارنکس ، حنجره ، تراشه ،ریه و یا اندامهای دیگر
• سرطان پاپیلاری با کانونهای تمایز سنگفرشی ( در 45-15 % کارسینومهای پاپیلری رخ میدهد )
سایر کارسینومهای تیروئید با تمایز سنگفرشی از جمله کارسینوم موکواپیدرموئید ، کارسینوم اسکلروزان موکواپیدرموئید با ائوزینوفیلی ، CASTLE
مشابه با سرطان آناپلاستیک سالمندان مبتلا به گواتر مزمن را تحتتاثیر قرار میدهد .
تظاهرات بالینی:
بیماران سالمند با رشد سریع توده گردنی مراجعه میکنند – بیماران با سابقهی طولانی بیماری تیروئید – متاستازهای خارج تیروئیدی و به گرههای گردنی متداول است اما متاستازهای دوردست نادر است .
پیشآگهی ضعیف با بقای متوسط 6 ماه – مرگ تقریبا در همه موارد که که معمولا ناشی از پیشرفت لوکال و تحت فشار قرارگرفتن مسیر هوایی است .
درمان : Radical resection و رادیوتراپی
Scc تیروئید
بسیار نادر ، بسیار کشنده و تماما از سلولهای سنگفرشی تشکیل شده است. از نظر بالینی و آسیبشناسی اشتراکاتی با سرطان آناپلاستیک تیروئید دارد و میتوان آن را به عنوان نوعی از سرطان آناپلاستیک تیروئید در نظر گرفت . مواردی که باید rule out شود :
• متاستاز یا تهاجم مستقیم از سلولهای سنگفرشی اوروفارنکس ، حنجره ، تراشه ،ریه و یا اندامهای دیگر
• سرطان پاپیلاری با کانونهای تمایز سنگفرشی ( در 45-15 % کارسینومهای پاپیلری رخ میدهد )
سایر کارسینومهای تیروئید با تمایز سنگفرشی از جمله کارسینوم موکواپیدرموئید ، کارسینوم اسکلروزان موکواپیدرموئید با ائوزینوفیلی ، CASTLE
مشابه با سرطان آناپلاستیک سالمندان مبتلا به گواتر مزمن را تحتتاثیر قرار میدهد .
تظاهرات بالینی:
بیماران سالمند با رشد سریع توده گردنی مراجعه میکنند – بیماران با سابقهی بیماری تیروئید – متاستازهای خارج تیروئیدی و به گرههای گردنی متداول است اما متاستازهای دوردست نادر است .
پیشآگهی ضعیف با بقای متوسط 6 ماه – مرگ تقریبا در همه موارد که که معمولا ناشی از پیشرفت لوکال و تحت فشار قرارگرفتن مسیر هوایی است .
درمان :
Radical resection و رادیوتراپی
تغییرات مولکولی در کارسینوم پاپیلاری تیروئید
اولین واصلی ترین تغییر قابل توجه درPTCبازآرایی کروموزومی RETیا جهش پروتوانکوژن های RASوBRAF است که باعث افزایش فعالیت پروتئین کینازهای میتوژن(MAPK) میشود.که سببافزایش تکثیر سلولی میشوند.
همچنینی مولکول هایی که به صورت فیزیولوژیک باعث رشد تیروسیت ها میشوندمانند اینترلوکین ۱و اینترلوکین۸ و همچنین سایر سایتو کاین ها(مانندIGF1,TGF beta,EGF)میتوانند در ایجاد این نوع از کنسر تیروئید نقشی ایفا نمایند.
افزایش بیان و تولید پروتئین هایی مانندheat shock protein70,,,proxiredoxin,,,isoform s100protein و همچنینی افزایش تولیدmicroRNAها نیز درPTC قابل مشاهده است
Scc تیروئید
بسیار نادر ، بسیار کشنده و تماما از سلولهای سنگفرشی تشکیل شده است. از نظر بالینی و آسیبشناسی اشتراکاتی با سرطان آناپلاستیک تیروئید دارد و میتوان آن را به عنوان نوعی از سرطان آناپلاستیک تیروئید در نظر گرفت . مواردی که باید rule out شود :
متاستاز یا تهاجم مستقیم از سلولهای سنگفرشی اوروفارنکس ، حنجره ، تراشه ،ریه و یا اندامهای دیگر
سرطان پاپیلاری با کانونهای تمایز سنگفرشی ( در 45-15 % کارسینومهای پاپیلری رخ میدهد )
سایر کارسینومهای تیروئید با تمایز سنگفرشی از جمله کارسینوم موکواپیدرموئید ، کارسینوم اسکلروزان موکواپیدرموئید با ائوزینوفیلی ، CASTLE
مشابه با سرطان آناپلاستیک سالمندان مبتلا به گواتر مزمن را تحتتاثیر قرار میدهد .
تظاهرات بالینی:
بیماران سالمند با رشد سریع توده گردنی مراجعه میکنند – بیماران با سابقهی طولانی بیماری تیروئید – متاستازهای خارج تیروئیدی و به گرههای گردنی متداول است اما متاستازهای دوردست نادر است .
پیشآگهی ضعیف با بقای متوسط 6 ماه – مرگ تقریبا در همه موارد که که معمولا ناشی از پیشرفت لوکال و تحت فشار قرارگرفتن مسیر هوایی است .
درمان:
radical resction و رادیوتراپی
پانکراتیت مزمن:
پانکراتیت مزمن معمولاً در پی حملات مکرر پانکراتیت حاد بروز میکند زیرا لوزالمعده در بین حملات کاملاً بهبود نمییابد. لوزالمعده در جریان این عارضه، به تدریج توانایی تولید آنزیمهای گوارشی و هورمونهای ضروری مانند انسولین را از دست میدهد. التهاب پانکراس ناشی از خودهضمی بافت آن توسط آنزیمهای پانکراس به ویژه تریپسین میباشد. علایم پانکراتیت ممکن است به صورت دردهای مداوم یا متناوب در ناحیه فوقانی شکم احساس شوند (علایم با غذاخوردن تشدید میشود و دورههای درد گاه یک هفته طول میکشد). تهوع، استفراغ، اتساع شکم، زردی (یرقان)، اسهال چرب و کاهش وزن.
پانکراتیت اتوایمیون
شیوع این تظاهر از بیماری در ژاپن ٠.٨٢ درصد در هر ۱۰۰ هزار نفر است که به صورت گسترده رو به افزایش است. باید دو فرم پانکراتیت اتوایمیون (AIP) را از هم افتراق دهیم.
اول که مرتبط با اصل نام دیگرش پانکراتیت اسکلروتیک لنفوسیتیک است. فرم دوم که مستقل ازIgG4-RD است، با عناوین AIP-Type2 ,Idiopathic duct centric chronic pancreatitis, و پانکراتیت گرانولوسیتیک اپیتلیالی شناخته می شود.
فرم دوم پانکراتیت نسبت به فرم اول بسیار ناشایع است و گاهی با سندرم روده ملتهب (IBD) همراه است
اکثر بیماران علاوه بر پانکراتیت تیپ ۱ AIP با سایر تظاهرات IgG4-RD همراه هستند مثل کلانژیت اسکلروزتیک، لنفادنوپاتی و یا درگیری غدد بزاقی و لاکریمال.
تشخیص افتراقیAIP1 از آدنوکارسینوم پانکراس بر اساس تظاهرات بالینی برخی اوقات بسیار مشکل است؛ یرقان بدون درد به عنوان مثال یک یافته بسیار شایع در هر دو بیماری است و اکثر بیماران AIP1 بدون در نظر گرفتن سرطان پانکراس، پروسه whipple را متحمل میشوند و همچنین سطح IgG4 افزایش یافته در هر دو بیماری دیده می شود.
یافتههای رادیولوژیک AIP1 شامل بزرگی منتشر پانکراس که به نام پانکراس سوسیسی شکل شناخته میشود و هالهای از ادم گسترده اطراف پانکراس است. هر دوی این ویژگیها در سی تی اسکن شکم به خوبی مشاهده میشود.
سلولهای Tcell-cytotoxic CD4+دارای مولکولی به نام F7( SLAMF7) که یک سیگنال فعال کننده لنفوسیت است، روی سطح خود دارند.
آدنوم هیپوفیز ایزوله خانوادگی (Familial isolated pituitary adenoma) و یا FIPA یک اختلال ارثی است که با رشد یک تومور غیر سرطانی در غده هیپوفیز (آدنوم هیپوفیز-pituitary adenoma) مشخص می شود.
تومورهای ایجاد شده در غده هیپوفیز، می توانند مقدار زیادی از یک یا چند هورمون را آزاد کنند، اگرچه بعضی از این تومورها، هورمون تولید نمی کنند (آدنوم های هیپوفیز غیر عملکردی-nonfunctioning pituitary adenomas). تومورهایی که در تولید هورمون ها اختلال ایجاد می کنند، معمولا از طریق هورمون های خاص تولید شده، تشخیص داده می شوند. پرولاکتینوما ها (Prolactinomas) شایع ترین تومور در FIPA هستند. این تومورها پرولاکتین (prolactin)، هورمون محرک تولید شیر در زنان را تولید می کنند.
علاوه بر این، نوع دیگری از تومور به نام سوماتوتروپینما (somatotropinoma) نیز در FIPA شایع است. این تومورها تولید کننده ی هورمون رشد (که سوماتوتروپین-Somatotropin نیز نامیده می شود)، می باشند که این هورمون باعث رشد بدن میشود. سوماتوتروپینما می تواند در کودکان و یا بزرگسالان منجر به افزایش قد (ژیگانتیسم-gigantism) شود، زیرا رشد استخوان های دراز دست و پا ادامه دارد.
جهش در ژن AIP، می تواند باعث ایجاد FIPA شود. عملکرد پروتئین تولید شده از این ژن به خوبی مشخص نیست اما به نظر می رسد به عنوان یک مهارکننده تومور (tumor suppressor) عمل می کند. مهار کننده های تومور به جلوگیری از رشد و تقسیم غیر قابل کنترل سلول ها، کمک می کنند.
ژنتیک:FIPA دارای الگوی وراثتی اتوزومال غالب است. در این الگوی وراثتی، یک نسخه از ژن تغییر یافته در هر سلول برای ایجاد اختلال کافی است. با این حال، تنها 20 الی 30 درصد افراد دارای جهش در ژن AIP دچار آدنوم هیپوفیز می شوند. این پدیده، که در اثر آن برخی از افراد دارای جهش، دچار علائم بارز یک اختلال نمی شوند، نفوذ پذیری ناقص (incomplete penetrance) نام دارد.

انسولينوما
از تومورهاي نادر شکمي بوده که با ترشح انسولين باعث علايم شديد هيپوگليسمي و گاهي مرگ بيمار مي گردد.
محل اين تومور در غده پانکراس و با ابعاد کوچک 1 تا 2 سانتيمتر بوده و بروز ترياد ويپل علايم هيپوگليسمي، قند خون زير 50mg/dl بر طرف شدن علايم هيپوگليسمي با تجويز وريدي قند) علامت تيپيک تشخيصي آن است. اين بيماران معمولا بدليل تغذيه مواد قندي فراوان، چاق بوده و به دليل بروز اختلالات رفتاري – کاهش سطح هوشياري يا اختلالات حافظه معمولا تا مدتي با تشخيص مشکلات روحي و رواني درمان مي شوند. تشخيص هر چه سريعتر اين بيماران مهم است زيرا هيپوگليسمي مي تواند باعث آسيبهاي مغزي و حتي مرگ شود. درمان قطعي اين تومور فقط با رزکسيون آن قابل انجام است. براى مهار آزاد شدن انسولين، ديازوکسيد تجويز مىشود. براى کارسينومهاى غيرقابل درمان سلول جزيرهاي، بهترين داروى شيمىدرمانى استرپتوزوسين است.
تیروئیدیت فیبروزان مولتی فوکال :
نوعی از تیروئیدیت مزمن با مراکز فیبروز ، که بسیاری از ان ها همراه با آتیپی غیر فعال فولیکول ها می باشد.
علت آن از لحاظ پاتولوژی نامشخص است اما ممکن است تظاهری از بیماری مرتبط با IgG4 باشد .
تیروئید به علت فیبروز ، سفت و ثابت ارزیابی میشود ، ممکن است باعث ایجاد فشار به ساختمان های مجاور گردن شود.
تشخیص از طریق بیوپسی باز صورت میگیرد .
درمان آن جراحی بوده اما داروی تاموکسیفن ممکن است مفید باشد .
کارسینوم مدولاری:(MTC)
از سلولهای پارافولیکولر(سلول C)منشا میگیرد.این سلولها گیرنده TSHندارند و ید را جذب نمیکنند.اگرسلولهای بدست امده از توده تیرویید کرایتریای بدخیمی داشت و به شکل پلی گونال (چندوجهی)بوده و در سیتوپلاسم ان ماده امیلویید صورتی رنگ دیده شد تشخیص MTCاست.2نوع دارد.sporadic, familial (فامیلیال میتواند همراه با MEN2باشد.)تشخیص با FNAمی باشد.
درمان:جراحی است شامل تیروییدکتومی توتال وکمورادیاسیون .به ئلیل برداشتن تیرویید باید لووتیروکسین تجویزشود.
این نوع تومور تمایل زیادی به متاستاز به ریه،استخوان و کبد دارد.
علائم و نشانه های ناشی از کارسینوم هیپوفیز :
1. فلج شدن حرکت چشم که سبب دوبینی یا تاری دید می گردد .
2. فقدان بینایی محیطی .
3. کوری ناگهانی .
4. سردرد .
5. سرگیجه .
6. فقدان هوشیاری .
اختلالات بینایی زمانی رخ می دهد که تومور بر روی عصب بینایی فشار وارد آورد . فقدان ناگهانی هوشیاری و حتی مرگ به دلیل خونریزی ناگهانی داخل تومور نیز ممکن است رخ بدهد .
فقدان هر نوع هورمون علامت خاص خود را ایجاد می کند که شامل موارد زیر خواهد بود :
1. تهوع
2. ضعف
3. کاهش یا افزایش بی دلیل وزن
4. قطع قاعدگی در خانم ها
5. ناتوانی جنسی به صورت اختلال در نعوظ
6. کاهش میل جنسی.
درمان تومورهای هیپوفیز :
جراحی : جراحی عموما از طریق استخوان اسفنوئید انجام شده و غده هیپوفیز برداشته می شود . گاهی براشتن تومور با روش کرانیوتومی انجام می شود . به این ترتیب که جراح از طریق ایجاد شکاف در جمجمه تومور را برمی دارد . این روش فقط برای تومورهای بزرگ و پیچیده انجام می شود . رادیو تراپی .
رادیوتراپی کان ون تیونال : از این روش در مواردی استفاده میشود که تومور پس از انجام جراحی مجددا عود کرده یاعلائم ناشی از آن با دارو تسکین نیابد . در این روش اشعه مستقیما به تومور تابانده می شود . این کار 5 بار در هفته و به مدت 4-6 هفته انجام می گیرد . رادیو تراپی استروتاکتیک : این یک روش جدید رادیوتراپی است که در آن از اشعه گاما استفاده می شود که مستقیما به تومور تابانده و بر روی آن متمرکز خواهد شد.
واریانت های شایع PTC:
Follicular Variant
این تومورها وقتی مورد بررسی قرار می گیرند ، مانند نئوپلاسم فولیکول هستند, از فولیکولهایی با اندازه متغیر تشکیل شده اند. کلوئید معمولاً در مقایسه با کلوئید در تیروئید غیر نئوپلاستی مجاور تیره یا hypereosinophilic است و ممکن است نمای”bubble gum” را نشان دهدسلول های غول پیکر چند هسته ای گاه به گاه در فولیکول ها وجود دارند
پیش آگهی این تومورها مشابه PTC معمولی است. یک استثناء نوع فولیکول پراکندگی یا مولتی مدولار است که دارای یک دوره بالینی تهاجمی تر است.پیش آگهی نوع فولیکولار PTC همچنین به این بستگی دارد که کاملاً محصور شده یا تهاجمی هستند
Papillary Microcarcinoma
این تومورها معمولاً به طور اتفاقی یافت می شوند و قطر آن کمتر از 1 سانتی متر است.
گاهی ممکن است بیماری همرا با متاستاز غدد لنفاوی گردن تظاهر یابد. تومورها اغلب نزدیک به کپسول تیروئید هستند. در صورت عدم محاصره بودن و وجود اسکلروز گسترده ممکن است نسبت به تومورهای کاملاً محصور شده تهاجمی تر باشد
Tall Cell Variant
این نوع توموراز سلولهایی تشکیل شده است که ارتفاعشان حداقل 2-3 برابر عرضشان است. سلولهای تومور سیتوپلاسم ائوزینوفیلی ویژگیهای هسته ای مشابه PTC معمولی دارند
Oncocytic Variant
این تومورها در معاینه شبیه به تومورهای سلول فولیکولی Hurthle هستند و از رنگ قهوه ای مشخصی برخوردار هستند. ممکن است نمای فولیکولی یا پاپیلاری داشته باشند
Columnar Cell Variant
این یک نوع نادر است که از سلولهای استوانه ای کاذب تشکیل شده است. برخی از سلولها ممکن است واکوئل های سیتوپلاسمی سوپرا نوکلئار و ساب نوکلئار داشته باشند
تیروئیدیت هاشیموتو:
این بیماری یک التهاب لنفوسیتیک اتوایمیون تیروئید است که در نتیجه ی تخریب سلول های فولیکولار تیروئید که در نهایت منجر به هایپو تیروئیدیسم میشود، رخ میدهد. تظاهر بالینی عمده ی آن بزرگ شدن منتشر و بدون درد تیروئید در زنان میانسال است. در تیروئید بیمار توده های ارتشاح لنفوسیتیک که اغلب همراه با فولیکول های حاوی مراکز زایا هستند دیده میشود، و هر دو نوع لنفوسیت های T cytotoxic و T helper افزایش پیدا میکنند. T cytotoxic باعث تخریب سلول های تیروئید میشوند و بعلاوه سلول های T به صورت موضعی سایتوکاین هایی مثل TNF ،IL1 ،INF ترشح میکنند که باعث آپوپتوز و از بین رفتن سلول های تیروئید میشود. آنتی بادی های علیه TPO و Tg در بیش از 90% موارد مثبت است که صحه ای بر اتوایمیون بودن بیماری است. در این بیماری درجات مختلفی از فیبروز مشاهده میشود.
HTA(Hyalinized Trabecular Adenoma)
این نئوپلاسم که با نام Hyalinized Trabecular Tumor هم شناخته می شود یک تومور نادر مشتق از سلول های فولیکولی غده تیروئید می باشد.
با توجه به ویژگی های هستهای مشابه و تغییرات ژن RET می توان آن را به نوعی یک کارسینوم پاپیلاری تیروئید محسوب کرد.(افتراق این دو از هم بسیار مشکل می باشد)
سن بروز بین ۲۱ تا ۸۰ سال متغیر بوده و زنان را به نسبت ۱.۶ به مردان مبتلا می کند. این بیماران عموماً بدون علامت هستند و شدت و نوع علائم آنها میتواند با توجه به اندازه و موقعیت آناتومیکی تومور متفاوت باشد.
بروز این آدنوم می تواند با التهاب لنفوسیتی تیروئید(تیروییدیت لنفوسیتی) و گواتر مولتی ندولر مرتبط باشد.
در اکثریت قریب به اتفاق موارد این تومور خوش خیم بوده ولی در مواردی تهاجم عروقی یا کپسولی نشان داده است.
نمای میکروسکوپی(Pathology)
●نمای ترابکولار در آن می تواند به شکل straight or curved pattern باشد
●دارای سلول های چند ضلعی با اندازه های متغیر که مرزهای سلولی در آن مشخص است.
●نمای کلیدی پاتولوژی آن سیتوپلاسم ائوزینوفیلی دارای ماده هیالن می باشد(فضای خارج سلولی هیالیننزه یا کلسیفیه شده)
●سیتوپلاسم حدود ۲ تا ۵ میکرون بوده و زرد و کروی می باشد.
●وجود شیار های هسته ای در نمای پاتولوژی بسیار متداول است.(Nulear grooves)
●ممکن است اجسام Psammona هم دیده شود.
جهت فهم بهتر slide های مربوط به نماهای پاتولوژیکی ذکر شده، آورده شده است.
■تشخیص اولیه به کمک سونوگرافی یا FNA بوده و درمان این بیماری لوبکتومی تیروئید می باشد.
پانکراس حلقوی یک بیماری نادر است که در آن قسمت دوم دوازدهه با حلقه ای از بافت لوزالمعده(عمدتا سر لورالمعده) به طور مداوم احاطه می شود. این قسمت از لوزالمعده می تواند دوازدهه را تنگ کند و جریان غذا را به سایر نقاط روده بلاک یا مختل سازد.
علائم اولیه بارز ناهنجاری شامل: پلی هیدرامانیوس (مایع اضافی مایع آمنیوتیک) ، وزن کم هنگام تولد و در مواردی همراه با استفراغ غیر صفراوی است (انسداد عموما بالای پاپیلای واتر است : بالاتر از محل اتصال مجاری صفراوی). بیماریهای مختلف کروموزومی (به عنوان مثال تریزومی 21 و با فرکانس جزئی ، تریزومی 18 و تریزومی 13) در حدود 33٪ از افراد مبتلا به پانکراس حلقوی وجود دارند.
در بزرگسالان: درد شکم در ناحیه اپیگاستریک ، حالت تهوع و استفراغ است که ممکن است قبل از رسیدن به یک تشخیص دقیق ، برای مدت طولانی (گاهی اوقات سالها) وجود داشته باشد.
این وضعیت خاص به طور معمول با رشد جنین شناسی غیرطبیعی همراه است ، اما میتواند در موارد بالغ و بعد از تولد نیز ایجاد شوند. این می تواند ناشی از :
1-رشد یک جوانه لوزالمعده شکمی در اطراف دوازدهه باشد ، جایی که قسمت هایی از جوانه شکمی با جوانه پشتی بهم متصل میشوند و یک حلقه لوزالمعده تشکیل می دهند. و یا اگر2- جوانه لوزالمعده شکمی نتواند بطور کامل چرخش کند ، بنابراین در سمت راست باقی می ماند یا اگر 3-جوانه پشتی در جهت اشتباه بچرخد ، به گونه ای که اثنی عشر توسط بافت لوزالمعده احاطه شود.
روشهای تشخیصی پس از زایمان شامل پرتونگاری و سونوگرافی شکمی ، سی تی اسکناست. رادیوگرافی شکمی می تواند نشانه کلاسیک “حباب مضاعف” را نشان دهد: وجود هوا در معده و دوازدهه. متأسفانه ، این علامت دو حبابی برای لوزالمعده حلقوی پاتولوژیک نیست ، زیرا می تواند در سایر شرایط ، مانند آترزی دوازدهه و ناهنجاری روده نیز مشاهده شود.
در نوزادان ، درمان برای تسکین انسداد معمولاً دئودنوژژونوستومی است.در بزرگسالان ، به دلیل تحرک جزئی اثنی عشر ، رویکرد گاستروژژونوستومی لاپاروسکوپی یا duodenojejunostomy است.
سودوسیست پانکراس
این عارضه بیشتر در کودکان ظاهر میشود.
علت:این عارضه معمولا بعد از پانکراتیت حاد رخ میدهد. هنگامی که توسط عفونت و یا التهاب، پانکراس تخریب شود، کیست ایجاد میشود.
بعد از آسیب به شکم و در برخی افراد با پانکراتیت مزمن، این عارضه ایجاد میشود.
علایم: نفخ شکم-
درد دائمی و یا درد شدید شکم که ممکن است به پشت هم کشیده شود-
مشکل در غذاخوردن و هضم کردن غذا
تشخیص:
احساس توده در سمت چپ و یا در وسط شکم- CT-scan -MRI – سونوگرافی
درمان: بستگی به اندازهی کیست در شکم دارد. کسیتهایی که بیش از ۶ هفته در شکم باقی میمانند و ضخامت آنها از ۵ سانتی متر بیشتر است، نیاز به جراحی دارند.
عوارض: آبسه پانکراس-ممکن است کیست پاره شود، در نتیجه بیمار دچار شوک و خونریزی بیش از حد خواهد شد.ممکن است فشار را به اندامهای دیگر منتقل سازد.
تیروئیدیت گرانولوماتوز تحت حاد ( De Quervain):
تیروئیدیت گرانولوماتوزتحت حاد به عنوان تیروئیدیت De Quervain یا تیروئیدیت سلول غول پیکر شناخته میشود .سن شایع ابتلا 30-50 سال بوده و در زنان بیشتر دیده میشود .در اکثر بیماران سابقه ابتلا به عفونت های تنفسی فوقانی وجود دارد علل ویروسی درگیر کننده شامل :ویروس Coxsackie،اوریون و آدنو ویروس ها است .
از نظرعلایم بالینی :آغاز بیماری حاد بوده و با درد گردن(هنگام بلع) ،تب ،بزرگی تیروئید ، ESR بالاو لکوسیتوز مشخص میشود .هیپرتیروئیدیسم گذرا (یعنی TSH ساپرس شده وهورمون T3,T4توتال و فری افزایش یافته ) به دنبال از هم گسیختگی فولیکول های تیروئید در برخی از افراد دیده میشود که با پیشرفت تخریب غده ممکن است به هیپوتیروئید تبدیل شود .تظاهرات بالینی در مرحله هایپرتیروئید میتواند مواردی از علایم Graves راتقلید کند.در چنین مواردی جذب و اسکن رادیونوکلئید میتواند مفید باشد زیرا تیروئیدیت تحت حاد باعث کاهش جذب ایزوتوپ و گریوزمنجر به افزایش جذب میشود .از طرفی بیماری گریوز را میتوان با رادیوتراپی درمان کرد اما تیروئیدیت تحت حاد معمولا خود محدود است و با رادیوتراپی درمان نمیشود .
از نظر پاتولوژی:گسیختگی فولیکولها و خروج کلوئید منجر به ایجاد واکنش التهابی گرانولومی و تجمع سلول های giant میشود. بارفع التهاب ،بافت فیبروز جایگزین خواهد شد.
درمان با استفاده از مسدودکننده های بتا ،آسپیرین و NSAID است (اگر جواب نداد از گلوکوکورتیکوئید استفاده میکنیم )
این وضعیت نوعا خود محدود است و در اغلب بیماران طی 6-8 به حال طبیعی باز می گردد.
پاپیلوم سینونازال (پاپیلوم اشنایدری)
این تومور خوش خیم بوده و منشا آن مخاط سینونازال است.
و میتواند بافت پوششی استوانه ای یا مطبق سنگفرشی داشته باشد.
اینگونه به نظر می آید که ویروس HPV تیپ ۶ و ۱۱ مسئول ایجاد آن میباشد.
انواع آن به ترتیب شیوع عبارتند از:
Exophytic و Inverted و سیلندری
نوع Inverted برخلاف دو نوع دیگر همانگونه که از اسمش مشخص است رشد پاپیلومایی به درون خود دارد.
علی رغم خوش خیم بودن میتواند کانونهایی از بدخیمی نیز داشته باشد و به صورت موضعی حمله به مخاط اربیت و جمجمه داشته باشد.
عود در این تومور بسیار بالاست.
اختلالات مولکولی در FTC
باز آرایی pax8/PPARy : جابه جایی کروموزومیt(2,3)[q13;p25] باعث الحاق دو ژن pax8 , PPARy می شود ژن pax8 عامل رونویسی را کد میکند که برای نمو تیروئید ضروری است و بیان ژن های اختصاصی تیروئید پروکسیداز و تیروگلوبولین را تحریک میکند ژن PPARy نیز در تمام بافت ها عامل رونویسی را کد میکند و باز آرایی این ژن در FTC نقش دارد
جهش های فعال کننده RAAS : این جهش ها هم در تومور های خوش خیم هم در بدخیم وجود دارد و به تنهایی نمیتواند باعث بدخیمی شود شاید وقوع این جهش ها یک رخداد اولیه در تومور زایی تیروئید است
جهش های پروموتر TERT: در ftc نسبت به ptc با فرکانس بالاتری وجود دارد
متیلاسیون: جهش یا حذف ژن pTEN تغییر ژنتیک کلاسیکی است که سبب فعالیت pi3K/AKT میشود و عامل تومور زایی در افراد مبتلا به این سندرم است متیلاسیون pTEN در سرطان های فولیکولار شایع است هایپر متیلاسیون RASSF1A که به عنوان سرکوبگر تومور شناخته میشود در ۷۵ درصد سرطان های فولیکولار نقش دارد
فعالیت تلومرازی از دیگر عوامل موثر در تهاجمی بودن سرطان تیروئید است افزایش فعالیت تلومراز ناشی از تغییرات بیان ژنی hTERT بررسی شده است
همچنین Mira_146b و Mir_183 ممکن است توسعه این نوع سرطان تیروئید را از طریق القای مهاجرت و مهار مرگ سلولی برنامه ریزی شده تحت تاثیر قرار دهد
سیده الهه راهنما
Riedel’s thyroiditis
تیروئیدیت ریدل, یک اختلال نادر با علت ناشناخته است که باعث فیبروز وسیع در تیرویید و نقاط دیگر بدن مثل مدیاستن و رتروپریتوئن میشود. فیبروز شدید و مهاجم در کپسول تیروئید به بافت های اطراف گسترش یافته و حتی میتواند به نای و مری و عضلات عروق گسترش یابد. علیرغم این تغییرات هیستولوژیک وسیع، اختلال عملکرد تیروئیدی شایع نیست.
از نظر بالینی خوشخیم است ولی اهمیت ان از این جهت است که غده تیروئید در این بیماری، سفت و غیرحساس (بدون تندرس)، و غالبا غیرقرینه و چسبیده و ثابت است که به دلیل چسبندگی به بافت های اطراف گردن,تقلید بدخیمی میکند و ممکن است با بدخیمی اشتباه گرفته شود.
تشخیص نیازمند بیوپسی باز است، چون بیوپسی FNA معمولا کافی نیست.
درمان این بیماری جراحی است و با هدف برطرف شدن نشانه های ناشی از فشار بر روی نای،مری و عروق و اعصاب گردن انجام میشود.(علائم فشاری :استریدور و خشونت صدا- تنگی نفس و دیسفاژی)
Undifferentiated carcinoma: تومور تمایزنیافته از منشا اپیتلیوم فولیکولی است وبسیار مهاجم است عمدتا از فقدان تومورهای دیگر در نتیجه یک یا چند تغییر ژنتیکی مثل جهش های نقطه ای غیر فعال کننده ژن سرکوبگر P53 ناشی میشوند .متوسط سن ابتلا 65 سال است .در بررسی ماکروسکوپیک توده حجیم با رشد سریع با تهاحم به کپسول و ساختارهای گردنی مشاهده میشود .در بررسی میکروسکوپیک نئوپلاسم متشکل از سلول های بسیار آناپلاستیک به صورت سلول های gaint ،سلول دوکی (نمای سارکوماتو)وترکیبی از دو نمای مزبور میباشد .
اگر بتواند ید رادیواکتیو را جذب کند میتوان از ید رادیواکتیو برای درمان سودجست و در صورتی که به رادیو تراپی با اشعه خارجی پاسخ دهد میتوان از این درمان استفاده کرد .پروگنوز بسیار ضعیف است ودر تمام موارد منجر به مرگ میشود .
تیروئیدیت حاد،یک آبسه یا سلولیت یا عفونت چرکی حاد تیروئید است.
عامل آن اغلب باکتریال است و بیمار با علائم سپسیس مراجعه میکند.
رادیاسیون ،درمان با بتا اینترفرون و آمیودارون هم از علل این بیماری هستند.
شایع ترین علت ان در اطفال و بالغین جوان ،وجود سینوس پیریفورم است، (باقی مانده ی بن بست حقی چهارم )که اغلب در سمت چپ است.
علایم بالینی شامل تب بالا،درد وتندرنس جلوی گردن تیر کشنده به گوش ،تورم یک طرفه تیروئید و دیسفاژی و اریتم روی غده است . علائم آزمایشگاهی شامل ESR بالا ،لکوسیتوز و افزایش IL_6 می باشد.
TFT نرمال (به علت عدم درگیری فولیکول ها) و اسکن تیروئید نرمال از ویژگی های تیروئیدیت حاد هستند.
تشخیص با کمک FAN ،نمونه گیری برای رنگ آمیزی و کشت صورت می گیرد .
درمان : میتوان آنتی بیوتیک تجویز کرد و اگر در CT یا سونوگرافی آبسه مشاهده شود لازم است که با جراحی آبسه را تخلیه کنیم .
پاپیلوم سینونازال ( پاپیلوم اشنایدری)
این تومور خوش خیم بوده و منشا آن مخاط سینونازال است.
و میتواند بافت پوششی استوانه یا مطبق سنگفرشی داشته باشد.
اینگونه به نظر می آید که ویروس HpV تیپ ۶ و ۱۱ مسئول ایجاد آن باشد.
انواع آن به ترتیب شیوع عبارتند از:
Exophytic و inverted و سیلندری
نوع inverted برخلاف دو نوع دیگر همانگونه که از اسمش پیداست ، رشد پاپیلومایی به درون خود دارد.
علی رغم خوش خیم بودن میتواند کانونهایی از بدخیمی داشته باشد و به صورت موضعی حمله به مخاط اربیت و جمجمه داشته باشد.
عود در این تومور بسیار بالاست.
تومور های متاستاتیک تیروئید:
اندامهای در خطر متاستاز سرطان تیروئید:
توده بدخیم تیروئید ممکن است به غدد لنفاوی، ریه ها، استخوان و گاهی مغز گسترش پیدا کند، در برخی شرایط سرطان تیروئید بسیار پیشرفته است و ممکن است به ساختار موجود در گردن مانند تراکه آ ، مری ، عروق خونی، اعصاب و یا عضلات منتقل شود، این حالت سرطان پیشرفته محل یا به اصطلاح لوکال گفته می شود.
درمان سرطان تیروئید مناستاتیک:
خوشبختانه اغلب متاستازها در ابتدا همچنان خاصیت جذب ید را دارند و به همین دلیل درمان ید رادیو اکتیو (RAI) همچنان به عنوان یک گزینه درمانی در نظر گرفته می شود، این درمان علی الخصوص برای سرطان های کوچک تیروئید با متاستاز ریه که در تصاویر اسکن نشان داده نشده اند ولی در اسکن رادیواکتیو مشخص شده است پاسخ گو خواهد بود.
متاستاز سرطان تیروئید در صورتی که پیشرفت کرده باشد و در سونوگرافی و یا CAT scan قابل مشاهده باشد، بایست تحت عمل جراحی و تیرئیدکتومی قرار بگیرد، در حقیقت جراحی روش معمول درمان متاستاز سرطان تیروئید و غدد لنفاوی در گردن است.
ریسک فاکتورها عبارتند از:
افراد بین ۲۵ تا ۶۵ سال زن بودن ، اگر به طریقی سر و گردن در معرض تابش اشعه قرار گرفته است و یا در محل بمب اتمی بودن (این عاملی است که می تواند تا ۵ سال بعد فرد را به سرطان مبتلا کند) ، داشتن گواتر و یا سابقه گواتر ، سابقه خانوادگی در بیماریها و یا سرطان تیروئید ، داشتن سوابق ژنتیکی از سرطان مانند بیماری های سرطان مدولاری تیروئید خانوادگی، سندروم نئوپلاست نوع ۲A غدد درون ریز چندگانه، سندروم نوع ۲B نئوپلاست غدد درون ریز چندگانه ،آسیایی بودن
برخی از علائم متاستاز تبروئید به اندام ها:
_متاستاز سرطان تیروئید به ریه:
سرفه به شکلی که قطع نشود ،تنگی نفس، عفونت مکرر قفسه سینه،سرفه های خونی، درد یا ناراحتی در قفسه سینه،کاهش وزن
_متاستاز سرطان تیروئید به استخوان :
درد استخوان،شکنندگی استخوان،مشکلات عصبی
ادنوم فولیکولار تیروئید:
ادنوم فولیکولار تیروئید یک تومور خوش خیم بدون کپسول غده ی تیروئید است و یکی از نئوپلاسم های شایع تیروئید میباشد و به دو گروه عملکردی و غیر عملکردی تقسیم بندی میشود که عملکردی همان ادنوم فولیکولار توکسیک است و وباعث هایپر تیروئیدی علامت دار میشود و 1%فولیکولار ادنوما هارا دربرمیگیرد.اتیولوژی:کمبود ید و گواتر اندمیک فاکتور های زمینه ساز کنسر های فولیکولار است و فعالیت انکوژن ها و موتاسیون های ژنی در ادنوم فولیکولار شایع است.علائم:یک ندول سفت و قابل لمس تیروئید در معاینه میباشد و عموما بدون علامت اند مگر اینکه توده بزرگتر باشد و علائمی مانند دیس پنه و خشونت صدا و درد گردن و دیس فاژی بروز کند.تشخیص با استفاده از اسپیراسیون سوزنی تیروئید و سونوگرافی و اندازه گیری سطح TSH سرم میباشد.درمان :شامل لوبکتومی تیروئید و Isthmusectomy بوده و در بیماران ادنوم فولیکولار عملکردی میتوان ید رادیواکتیو 131 تجویز کرد و نیاز به لوبکتومی نیست.در سیر بیماری ادنوم فولیکولار غیر عملکردی ممکن است به کارسینوما تبدیل شود.
PTCشایعترین نوع سرطان تیروئید است. با اینکه رشد آن کند است ولی باز هم در صورت عدم درمان امکان پیشرفت دارد
این سرطان از سلول های تیروئید آغاز و ممکن است بجز تیروئید به غدد لنفاوی یا ریه کبد و مغز استخوان نیز سرایت کند
علایم: در اکثر موارد بدون علامت است ولی معمول ترین علامت آن وجود توده در گردن به صورت توده جامد نامنظم و یا چیتیک است که از رشد غیر طبیعی بافت به وجود می آید
سن رایج این سرطان بین ۳۰ تا ۵۰ سال است و در زنان شایع تر است درمان بستگی به زمان تشخیص دارد اگر توده از یک و نیم سانتی متر کمتر باشد درمان با پیش آگهی خوب امکان پذیر است و زیر ۵۵ سال به درمان بهتر پاسخ میدهند
درمان اولویت دار جراحی تیروئید است
اگر توده کوچک تر از یک سانتیمتر باشد یا به هر دلیلی امکان جراحی نباشد یا با وجود جراحی دچار شده باشد می توان از درمان kf تیروئید یا مایکروویو استفاده کرد
تشخیص :FNA سونوگرافی داپلر،mri،ct تیروئید برای بررسی میزان هورمون ها و فاکتورهای خون
چیتیک=سیستیک
درمان kf=درمان RF
عذرخواهی میکنم اصلاح کنید?
بیماری پانکراتیت حاد
فعال سازی نامتناسب پیش ساز های آنزیم های پانکراسی , درون پانکراس باعث خود هضمی ,نکروز , ادم و خونریزی می شود.
شایع ترین علت آن مصرف زیاد الکل یا سنگه های صفراوی و بیماری های صفراوی می باشد .
نشانه های پانکراتیت حاد شامل ۱_ درد شدید و ثابت اپیگاستریک که یک الی چهار ساعت پس از صرف غذا یا مصرف الکل زیاد آغاز می شود ۲- کاهش درد به هنگام خم شدن به جلو ۳ – تهوع و استفراغ ۴-انتشار درد به پشت ۵ -کبودی پهلوها و اطراف ناف…
تشخیص پانکراتیت حاد :
۱.ام آر آی و سی تی مثبت به همراه آمیلاز سرمی بالا تسخیص دهنده است .
۲.آمیلاز بالای ۱۰۰۰ نشان دهنده ی بیماری های صفراوی می باشد.
۳.نسبت آمیلاز به کراتینین ممکن است حساسیت تشخیص را افزایش دهد .
۴.افزایش LFT
۵.انجام ERCP
درمان پانکراتیت حاد:
۱.انجام NPO
۲. مایع درمانی
۳.آنتی بیوتیک ها
۴.ساکشن نازوگاستریک
۵.درمان هایپر گلایسمی و هایپو کلسمی همراه.
پیش آگهی و سیر بالینی:
موارد خفیف در مدت یک هفته برطرف می شوند .
موارد شدید ممکن است باعث شوک و سپسیس یا حتی مرگ شوند.
معیار های رانسون برای ارزیابی شدت بیماری:
اگر کمتر از ۳ معیار وجود داشته باشد میرایی کم تر از ۱ درصد و اگر معیار ها بیش از ۶ عدد باشد میرایی تا حد ۱۰۰ درصد افزایش می یابد..
ممکن است کیست و آبسه پانکراس ۳ تا ۶ هفته بعد ایجاد شود .
ممکن است که پانکراتیت حاد به فرم مزمن در آید.
پانکراس دوقسمتیPancreas divisum)):
تعریف و اپیدمیولوژی:
شایع ترین تغییر شکل آناتومیک مادر زادی پانکراس انسان می باشد که در آن در دوران جنینی نیمه های شکمی و پشتی پانکراس بهم نمی چسبند. بدین ترتیب درناژ پانکراس از طریق پاپیلا های فرعی صورت می گیرد و در واقع در این ناهنجاری یک مجرای منفرد پانکراسی نداریم و اغلب در عوض آن دو مجرای مجزای پشتی و شکمی داریم. این اختلال در 7-10 درصد از جمعیت دیده می شود.
علایم بالینی و تشخیص:
در اکثر موارد ، بیماران بدون علامت هستند اما در بعضی موارد علایمی مانند درد شکم ، تهوع و استفراغ بروز می یابد . این آنومالی در اکثر بیماران آن هارا مستعد ابتلا به پانکراتیت نمی کند، اما ترکیبی از پانکراس دو شاخه و سوراخ فرعی کوچک می تواند باعث انسداد مجرای پشتی شود و فرد را مستعد ابتلا به پانکراتیت حاد و مزمن کند.
تشخیص با مشاهده وجود دو مجرای فرعی مجزا در پانکراس با استفاده از تکنیک های MRCP Magnetic Resonance) Cholangiopancreatography) و یا ERCP((Endoscopic Retrograde Cholangiopancreatography مسجل می شود .
درمان:
اگر با علایم بالینی همراه نباشد نیاز به مداخله ای نیست . اما در صورت بروز علایم بالینی مداخله اندوسکوپیک یا جراحی اندیکاسیون پیدا می کند.
سومین نوع توموری که در پانکراس ایجاد می شود گلوکاگونوما (به انگلیسی: Glucagonoma) یک تومور نادر سلولهای آلفای لوزالمعده است که منجر به تولید بیش از حد هورمون گلوکاگون میشود. تومورهای سلولهای آلفا معمولاً همراه با سندرم گلوکاگونوما دیده میشوند
علائم و نشانهها
تولید بیش از حد هورمون گلوکاگون منجر به افزایش قند خون از طریق فعال شدن فرایندهای آنابولیک و کاتابولیک بدن، از جمله گلوکونئوژنز و تجزیه و تحلیل چربی میشود. گلوکونئوژنز تولید گلوکز از پروتئین و اسیدهای آمینه میباشد. تجزیه و تحلیل چربیها نیز افزایش مییابد. نتیجه این میشود که سطح بالایی از گلوکاگون، کاهش سطح خونی اسیدهای آمینه و کاهش تعداد گلبولهای قرمز خون همراه با اسهال و کاهش وزن تا ۱۵ کیلوگرم را در بیمار میشود مشاهده کرد.
راش (لکه پوستی) در نواحی مختلف بدن، زخم زبان
علت
اگر چه علت این بیماری ناشناخته است، عوامل ژنتیکی نقش مهمی در برخی از موارد بازی میکند. سابقه خانوادگی نئوپلازی متعدد نوع یک غدد درون ریز میتواند عامل خطر باشد. این تومورها معمولاً بدخیم هستند.
تشخیص
سرم خونی گلوکاگون با غلظت ۱۰۰۰ pg./میلی لیتر یا بیشتر نشان دهنده گلوکاگونوما میباشد. (محدوده نرمال بین ۵۰ تا 200 pg/mL میباشد.
در CBC میتوان علائم کم خونی را مشاهده کرد.
لکه پوستی در تشخیص گلوکوناما از اهمیت فراوانی برخوردار است.
موقعیت تومور را میتوان به وسیله ابزارهای رادیوگرافی از جمله آنژیوگرافی، سی تی اسکن، ام آر آی یا پت اسکن مشخص نمود.
درمان
تنها راه درمان برای گلوکاگونوما جراحی و برداشتن کامل تومور ست.
Pituicytoma توموري نادر در لوب خلفی هيپوفيز است که از سلولهای بزرگ كف الود تشکیل شده است و همچنين به ان infundibuloma مي گوييم. این موارد به ندرت در طول زندگی بيمار مشخص می شوند و اغلب اوقات در کالبد شکافی و MRI كه به علت هاي ديگر انجام مي شود كشف مي شوند.این تومورها ناشی از سلول هاي pituicytes هستند که سلولهای استرومائی (پشتیبان) لوب خلفی هستند. آنها ممکن است با نقص میدان بینایی ، سردرد ، كم كاري هيپوفيز و یا در طول زندگی بدون علامت باشند آنها معمولاً برخلاف موقعیت مکانی اي که دارند باعث ديابت بي مزه نمي شوند. به نظر می رسد که جراحی روش درمانی مطلوب است اما از آنجا که بسیار نادر هستند ، درمان قطعي براي ان ها و جود ندارد. آنها نسبت به آدنوم هاي استاندارد هیپوفیز كه ناشی از لوب قدامی غده هستند تمايل عروقی بيشتري دارند و به اين دليل جراحی ان ها سخت تر است.
Pituicytoma توموري نادر در لوب خلفی هيپوفيز است که از سلولهای بزرگ كف الود تشکیل شده است و همچنين به ان infundibuloma مي گوييم. این موارد به ندرت در طول زندگی بيمار مشخص می شوند و اغلب اوقات در کالبد شکافی و MRI كه به علت هاي ديگر انجام مي شود كشف مي شوند.این تومورها ناشی از سلول هاي pituicytes هستند که سلولهای استرومائی (پشتیبان) لوب خلفی هستند. آنها ممکن است با نقص میدان بینایی ، سردرد ، كم كاري هيپوفيز و یا در طول زندگی بدون علامت باشند آنها معمولاً برخلاف موقعیت مکانی اي که دارند باعث ديابت بي مزه نمي شوند. به نظر می رسد که جراحی روش درمانی مطلوب است اما از آنجا که بسیار نادر هستند ، درمان قطعي براي ان ها و جود ندارد. آنها نسبت به آدنوم هاي استاندارد هیپوفیز كه ناشی از لوب قدامی غده هستند تمايل عروقی بيشتري دارند و به اين دليل جراحی ان ها سخت تر است
clear cell tumor
clear cellدر پاتولوژی به منظور دیدن سلول هایی است که سیتوپلاسم آنها در رنگ آمیزی H&E به صورت شفاف دیده میشوند.clear cell ها در واقع سلول هایی با قابلیت ترشحی در اپیتلیوم هستند و یکی از اجزای غدد عرقی نیز محسوب میشوند.
احتمال بروز این درگیری به طور شایع در 70 – 80 درصد کارسینوماهای کلیه و همچنین در تخمدان و بافت نرم و پوست نیز وجود دارد.
در کلیه به صورت افزایش ناگهانی در تعداد سلول ها تظاهر میابد که منجر به تشکیل mass(توده) میشود.دلیل اصلی آن ناشناخته میباشد اما عواملی از قبیل سیگار ، بعضی داروها و بیماری های ارثی مانند ون هیپل لیندو موثر اند.
renal clear cell carcinoma میتواند به صورت فامیلیال(5%) و اسپورادیک(95%) رخ دهد که به دلیل اختلال در بازوی کوتاه کروموزوم 3 رخ میدهد.در صورتی که به علت ون هیپل لیندو (VHL) باشد اختلال در 3p25 از کروموزوم 3 میباشد.
برای درمان در خط اول جراحی را خواهیم داشت در صورتی که نتوان به طور کامل توده را خارج کنیم برای FOLLOW UP از مواردی چون رادیوتراپی و کموتراپی استفاده میکنیم
متاستاز سرطان تیروئید به غددلنفاوی
گسترش سرطان تیروئید به غدد لنفاوی می تواند به دو شکل نمود داشته باشد، در حالت ماکروسکوپی غدد لنفاوی دچار تورم شده و بزرگتر از حد معمول هستند و تورم دیده می شود، در حالت میکروسکوپیک لنف نود ها در سایز تغییری نمی کنند و تنها در بررسی های میکروسکوپیک مشخص می شوند، نرخ عود مجدد بیماری در دو حالت متفاوت است.
البته وجود یک توده در گردن نشانه سرطان نیست بلکه یک هشدار برای بررسی بیشتر است ولیکن علائمی نیز ممکن است بیمار را درگیر نماید این علائم عبارتند از تب، لرز، تعیق شبانه، خستگی و کاهش وزن، همچنین وجود گره های لنفاوی بزرگ در بدن
درمان
با وجود عوارض زیاد هنوز هم خط اول در درمان سرطان تیروئید جراحی است ولیکن، به تازگی پزشکان به روشهای کمتر تهاجمی مانند آر افو ماکروویو پی برده اند .
از روش آر اف در موارد زیر استفاده می شود:
• به علت عود مکرر چندین مرتبه جراحی شده و به دلیل چسبندگی و فیبروز امکان عمل جراحی وجود ندارد
• غدد در نواحی هستند که دسترسی جراح به آنها بسیار مشکل است مانند زون ۶
• و یا اینکه غدد لنفاوی بدخیم زیر ۱۰ میلی متر هستند و ممکن است جراح آن ها را در حین جراحی پیدا نکند، این موارد از جکله مواردی هستند که برای آنها درمان با آر اف در نظر گرفته می شود.
تیروئیدیت لمسی یا palpation thyroiditis
واژه palpation به معنای تخمین سختی بافت بیمار با استفاده از دستان متخصص است.
گسترش التهاب در تیروئید در اثر اسیب مکانیکی به فولیکول های تیروئید میباشد.این اتفاق ممکن است در اثر پالپیشن مکرر و شدید تیروئید یا مداخله جراحی مثل دایسکشن رادیکال گردن بیوفتد.(یعنی خارج کردن گره های لنفاوی جهت بررسی متاستاز)
نوعی تیروئیدیت تحت حاد است .*در تیروئیدیت تحت حاد، در غده تیروئید یک ارتشاح یا اینفیلتراسیون التهابی مشخص تکه ای در نقاط مختلف دیده می شود که با تخریب فولیکول های تیروئید و گلبول های سفید بزرگ چند هسته ای در داخل بعضی از فولیکول ها همراه است. تغییرات فولیکولی به سمت تشکیل گرانولوم پیش رفته و در نهایت منجر به فیبروز می شوند.
علائم تیروئیدیت تحت حاد: در تیروئیدیت تحت حاد، بیمار معمولا با یک تیروئید بزرگ و دردناک مراجعه می کند و از درد گردن شاکی است. گاهی تب نیز دیده می شود.تشخیص بیماری بر اساس موارد زیر صورت میگیرد:علائم و نشانه های بیماری: دردگلو با انتشار به گوش و فک، حساسیت در لمس، گواتر، علائم تیروتوکسیکوز یا کم کاری تیروئید.
آزمایش ها: ESR بالا و لکوسیتوز و معمولا منفی بودن Anti TPO و آزمایش تیروئید (T3 ، T4 ، TSH).
در نهایت در صورت شک به تشخیص می توان از FNA و جذب ید رادیواکتیو (اسکن تیروئید) استفاده کرد. در FNA تیروئیدیت تحت حاد، سلول های بزرگ چند هسته ای دیده می شود و در اسکن تیروئید معمولا جذب ید رادیواکتیو کاهش یافته است.
*پاتولوژی تیروئیدیت لمسی multifocal granulomatous folliculitisرا نشان میدهد. سلولهای T در مقایسه با سلولهای B غالب هستند. ممکن است پرکاری تیروئید زودگذر به دلیل نشت هورمون پیش ساخته تیروئید در خون وجود داشته باشد.
کارسیونوم مدولاری تیروئید فامیلی (FMTC)
FMTC بیماری ارثی آتوزومی غالب می باشد. بر اثر جهش در پروتوانکوژن RET ایجاد میشود.
کارسینوم مدولاری تیروئید در 70% موارد تک گیر و در 30% باقی مانده یه صورت خانوادگی و در زمینه ی سندرم نئوپلاسم های متعدد اندوکرین (MEN) 2A یا 2B یا کارسینوم مدولاری تیروئید خانوادگی (FMTC) بدون همراهی با سندرم MEN روی می دهد. هر دو شکل خانوادگی و تگ گیر دارای جهش های فعال کننده ی RET می باشند. کارسیونوم مدولاری تک گیر و موارد خانودگی بدون ارتباط با سندرم MEN در بالغین دیده می شود و حداکثر میزان بروز آن دهه های پنج و شش عمر می باشد. برعکس، مواردی که همراه MEN-2A و MEN-2B هستند در سنین پایین تر و حتی کودکان دیده می شوند.
تومور MTC نئوپلاسم هایی نورواندوکرین هستند که از سلولهای C یا پارافولیکولار تیروئید منشاء میگیرد. سلولهای کارسینوم مدولاری تیروئید، اغلب قوام سفت و بدون کپسول دارند و هورمون کلسیتونین ترشح میکنند.
ریخت شناسی :
کارسینوم های مدولاری ممکن است به صورت ندول های منفرد یا ضایعات متعدد باشند که هر دو لوب تیروئید را گرفتار کرده اند.
** نمای مشخصه ی این تومور ها رسوبات آمیلوئید که از مولکول های کلسیتونین تغییر شکل یافته تشکیل شده اند می باشد.
** ویژگی خاص کارسینوم مدولاری خانوادگی حضور هیپرپلازی چند کانونی سلول C در پارانشیم تیروئی مجاور است. این ویژگی معمولا در تک گیر دیده نمی شود. به نظر می رسد که کانون های هیپرپلازی سلول C به عنوان ضایعات پیش ساز کارسینوم مدولاری باشند. تشخیص پاتولوژی با رنگ آمیزی ایمونوهیستوشیمیایی کلسیتوئین و مشاهده آن درون سیتوپلاسم سلول های تومور و در آمیلوئید استرومایی انجام می شود.
تظاهرات بالینی:
• ندول سفت یا توده در تیروئید.
• بزرگی غدد لنفاوی گردن.
• اغلب توده های گردنی دردناک هستند.
• گاهی 2 طرف تیروئید درگیر میشود.
• اغلب در 3/2 فوقانی غده تیروئید دیده شوند.
گواتر دیس هورمونوژنتیک
بزرگ شدن تیروئید به دلیل نقص های مختلف ارثی در هورمون تیروئید است که عدم گردش هورمون تیروئید موجب ترشح tsh میشود که این امر موجب تخریب بیش از حد و هایپرپلازی تیروئید میشود و علت نقص در ژن های رمز کننده انزیم هایی است که در سنتز هورمون های تیروئیدی نقش دارند بیشتر در کودکان است . در دوران نوزادی با گواتر مشهود و کم کاری مادرزادی تیروئید اختلال در رشد و ناتوانی ذهنی همراه است در بزرگسالی گواتر و شواهد کمتری از اختلال عملکرد تیروئید وجود دارد
تشخیص: سابقه خانوادگی- غربالگری های روتین نوزادان در رارزیابی رادیولوژیک شامل سیتی و سونو و اندازه گیری tsh درمایع امنیوتیک در رحم میتوان تشخیص داد
درمان: جایگزین t4تا زمانی که tshنرمال شود -بعضی با ید درمان میشوند- در انواع بزرگ جراحی- و در گواتر جنینی تزریق تیروکسین ب مایع امنیوتیک
تومور هرتل سل يا اونكوسايتوما
يكي از انواع كارسينوماي فوليكولار تيروئيد است،سلول هاي اكسيفيليك در تيروئيد اغلب هرتل سل ناميده ميشوند.در اين تومور سلول هاي اكسي فيليك دچار افزايش سايز با گرانول هاي سيتوپلاسمي ائوزينوفيلي مشخص ميشوند.
اغلب اين تومور با تيروئيديت هاشيموتو همراه است .
انواع مختلفي از خوش خيم تا بدخيم ميتواند داشته باشد،اما معمولا خوش خيم است و بعد جراحي عود نميكند.
علت مشخصي ندارد اما ميتوا رادياسيون سر يا گردن ،كمبود يد،موتاسيون ژنتيكي يا سابقه سرطان فاميلي را به عنوان محرك در نظر گرفت.
بيشتر درسنين بالاو ٧٠ تا ٨٠ سال ديده ميشود.
پانکراس هتروتروفیک یک بیماری نسبتاً نادر است . میزان بروز آن در مجموعه کالبد شکافی 0.5-13.7٪ است.
شایع ترین محل آن معده (38/25 درصد) و بعد آن دوازدهه (36/17 درصد) ، ژژنوم (15.21.7 درصد) و بندرت در مری ، مجرای صفراوی مشترک ، طحال ، مزنتری ، مدیاستن و لوله فالوپ است. در مورد پاتوژنز هتروتوپی پانکراس نظریه های مختلفی وجود دارد. دو فرضیه پیشنهادی محتمل که در مورد بافت پانکراس هتروتروفیک توضیح می دهند عبارتند از: 1) جوانه های بافت پانکراس جنینی جدا شده در حین چرخش foregut و همجوشی قسمت پشتی و شکمی لوزالمعده متعاقباً به طور مستقل به عنوان جزایر بافت پانکراس در دستگاه GI رشد می کند. 2) یک استعمال نامناسب لوزالمعده از بافت آندودرمال هنگام جنین زایی
نوع اول از بافت پانکراس معمولی با سلولهای acini ، مجاری و جزایر تشکیل شده است. نوع II مجرای لوزالمعده را نشان می دهد. نوع III که فقط از بافت آکینار تشکیل شده است. نوع IV تنها جزایر (پانکراس غدد درون ریز) است.
لوزالمعده هتروتروفیک معمولاً بدون علامت است. درد شایعترین علامت است و علائم دیگر ممکن است به دلیل اثر انسداد مانند انسداد روده معده و زردی انسدادی ایجاد شود. عوارض نادر شامل خونریزی ، پانکراتیت ، تصورات درونی ، انسولینوما ، سندرم زولینجر-الیسون ، کیست شبه پانکراس آبسه لوزالمعده و تحول بدخیم در بافت لوزالمعده هتروتروفیک است.
تشخيص افتراقي شامل تومور استرومايي دستگاه گوارش (GIST) ، تومور عصب اتونوم دستگاه گوارش (GANT) ، کارسينوئيد ، لنفوم و آدنوکارسينوما است [7]. در آندوسکوپی ، یک ضایعه زیر مخاطی عمود بر پایه ذکر شده است. سونوگرافی آندوسکوپی (EUS) یک عامل تشخیصی مفید در شناسایی ضایعه محسوب می شود. EUS و CT اسکن همزمان یک روش برتر است. بیوپسی معمول آندوسکوپی ممکن است سطحی باشد و منجر به نتیجه نادرست شود. با این حال ، بیوپسی جامبو در بسیاری موارد می تواند کمک کننده باشد. بیوپسی منجمد از عمل جراحی می تواند این تشخیص را ایجاد کند. ضایعات علامتی و مشکوک برای برداشتن جراحی در نظر گرفته می شوند. .
بیماری Graves یک اختلال سیستم ایمنی است که سبب تولید بیش از حد هورمون هلی نیرویی (هایپرتیروییدیسم) می شود. گرچه اختلالات مختلفی می تواند به هایپرتیروئیدیسم منجر شوند، بیماری Graves یک علت شایع آن است.
این بیماری هرفرد را می تواند مبتلا کند اما در خانم ها و در سن زیر ۴۰ سال شایع تر است.
مکانیسم این بیماری افزایش آنتی بادی محرک گیرنده های تیروئید(TSI) است که مشابه TSH(هورمون محرک تیروئید) عمل می کند.
دو مشخصه بارز آن افتالموپاتی و درماتوپاتی است که با مشاهده آن ها می توانیم به تشخیص برسیم. علایم بالینی آن عبارت اند از: بی قراری و عدم تحمل به گرما، ترمور، تاکی کاردی، کاهش وزن، بزرگ شدن غده تیروئید (گواتر)، اگزوفتالمی و… .
در این بیماری سطح هورمون T3 و T4 افزایش و هورمون محرک تیروئید (TSH) کاهش پیدا می کند.
برای درمان از داروهایی مانند متیل مازول، پروپیل تیواوراسیل و کاربیمازول استفاده می شود.
Pituitary hyperplasia هیپرپلازی هیپوفیز : افزایش غیر نئوپلاستیک در تعداد سلول های آدنوهیپوفیز( هیپوفیز قدامی )
ویژگی های ضروری: هیپرپلازی ندولار بدون فشار آوردن به بقیه نواحی یا یک افزایش منتشر سلول های هیپوفیزی بدون از بین رفتن ساختار غده
اپیدمیولوژی: به طور معمول بسیار نادر در نظر گرفته می شوند اما تحقیقات کالبد شکافی شیوع را به میزان 26% گزارش می دهد و ممکت است در هایپرپلازی پرولاکتین در بارداری شایع تر باشد. به طور معمول در بالغین جوان مشاهده می شود.
محل : آدنوهیپوفیز
پاتوفیزیولوژی:
پاسخ فیزیولوژیک به یک از کار افتادگی عضو انتهایی:
*شایع ترین » هیپرپلازی پرولاکتین در بارداری ، شیردهی یا استروژن درمانی
*هیپرپلازی سلول های ACTH به دلیل کمبود کورتیزول در بیماری آدیسون
*هیپرپلازی سلول های TSH همراه با هیپوتروئیدیسم اولیه طولانی مدت
*هایپرپلازی گنادوتروپ در هیپوگنادیسم اولیه به دلیل سندروم های کلاین فلتر و ترنر
هایپرپلازی پاتولوژیک همراه با هورمون های آزادکننده زیاد ترشح شده از جایی غیر از هیپوتالاموس:
*هایپر پلازی سلول های هورمون رشد به دلیل افزایش هورمون آزاد کننده هورمون رشد ترشح شده توسط تومور سلول های جزایر پانکراس ، فئوسیتوکروما، تومور های کارسینوئید و برونشیال
*هایپرپللازی سلول های ACTH به دلیل ترشح هورمون آزادکننده کورتیکوتروپین از هامارتوم هیپوتالاموس یا تومور های نورواندوکرین
ایدیوپاتیک
ارثی
ویژگی های بالینی:
هایپرپلازی های مرتبط با هورمون
سلول های هورمون رشد: غول آسایی یا آکرومگالی
سلول های پرولاکتین: hyperprolactinemia هایپر پرولاکتینمی
سلول های ACTH : سندرم کوشینگ
سلول های TSH : hyperprolactinemia
کم کاری تیروئید اولیه طولانی مدت منجر به هایپرپلازی TSH می شود
عدم وجود بازخورد منفی از تیروکسین (T4) منجر به افزایش سطح TRH می شود که سلولهای TSH و PRL هیپوفیز را تحریک می کند.
LH-FSH : نتایج اولیه شروع هیپوگنادیسم
تشخیص
بیوپسی
به ندرت با جراحی خارج می شود
به احتمال بسیار زیاد در کالبد شکافی شناسایی می شود
درمان :
معمولا اختلال زمینه ای اندوکرینولوژیک را باید تصحیح کرد
جراحی : برای هایپرپلازی سلول های هورمون رشد و ACTH
درمان کمکی : دارو درمانی برای کم کاری تیروئید
آدنوم هیپوفیزی (pituitory adenoma)
آدنوم های هیپوفیزی توموهای نورواندوکرین هیپوفیز قدامی هستند که از سلول های هیپوفیزی ترشح کننده هورمون تشکیل شده اند. تیپ های اصلی این تومورها شامل سوماتوتروپ، لاکتوتروپ، تیروتروپ، کورتیکوتروپ، گنادوتروپ، سلول های خنثی (Null) و چند هورمون (plurihormonal) هستند. آدنوم های هیپوفیزی 10 تا 15% نئوپلاسم های داخل جمجه را تشکیل می دهند. شیوع آن ها در دهه ی چهارم تا هفتم زندگی است. میزان شیوع اندکی در زنان بیشتر است. اکثر این تومورها در زین ترکی و درون لوب قدامی هیپوفیز یافت می شوند، گرچه موارد نادر در ساقه هیپوفیزی و حتی به عنوان بخشی از تراتوماهای تخمدانی نیز گزارش شده اند. اکثر بیماران (میکروآدنوماها) تظاهرات افزایش هورمون را نشان می دهند. تومورهای بزرگ تر (ماکروآدنوماها، اندازه بیش از 1 سانتی متر) اثراتی همچون سردرد یا اختلال در بینایی نیز ایجاد می کنند. نکروز هموارژیک تورموهای بزرگ یک اورژانس جراحی تلقی می شود. در بررسی های آزمایشگاهی اندازه گیری سطح پرولاکتین سرم، ACTH سرم، هورمون های تیروئیدی و… برای تشخیص استفاده می شود. در بررسی های میکروسکوپی باید یافته های بافت شناسی معمول مثل درجه میتوز، سلول های ژانت، انکولوزیون ها، خونریزی و ویژگی های عروقی مد نظر قرار گیرد. یکی از راه های درمان این تومورها جراحی است.
۵ سلول موجوددرهیپوفیز قدامی:
1 .سوماتوتروپ ها 30 تا 40 درصد
2 .کورتیکوتروپ ها 20 درصد
3 .تایر توپها 3 درصد
4 .گنادوتروپ و لاکتوتروپ ها
یکی از تظاهرات بالینی توده های هیپوفیز،Hemianopsia Hemolimusاست که بخاطر اثرات فشاری توده ایجاد میگردد. در ناحیه تمپورال نیز ممکن است آسیب ایجاد کند و فرد شنوایی خود را ازدست بدهد. که اثرات بستگی به سایز توده دارد.
توده هایی که درهیپوفیزترشح دارند:
.1آدنوم هایی که ترشح کننده هورمون هستند
ممکن است پرولاکتین ترشح کنند که منجر به آمنوره، گاالکتوره می شود.
هورمون رشد:اگر قبل از بلوغ باشدمنجر به ژیگانتیسم
اگر بعداز بلوغ باشد تبدیل به آکرومگالی می شود.
کورتیزول ایجاد کوشینگ و سندروم nelsonمی کند
TSH هایپرتیروئیدیسم را سبب می شود.
2 .ممکن است آدنوم هایی داشته باشیم که هیچ گونه ترشح هورمونی ندارد که 20 تا 25 درصد بیماران از این دسته هستند
معمولا علامتی برای بیماران ندارند و زمانی به تشخیص میرسیم که سایز توده بزرگ شود و مشکالت زیادی برای بیمار ایجاد کند زمانی علامت دار می شود که اثرات فشاری دارند یعنی زیاد بزرگ می شود و منجر به اختلالات بینایی سردرد و کمبود های هورمونی می گردد.
ناحیه ی پشتی بن بست سوم به غده ی پاراتیروئید تحتانی تمایز می یابد. ناحیه ی شکمی بن بست سوم هم تیموس را می سازد.
بخش پشتی بن بست چهارم غدد پاراتیروئید فوقانی را می سازد. بخش شکمی بن بست چهارم جسم اولتیموبرونشیال را می سازد که بعدا به سلول های c تیروئید که مسئول ساخت کلسی تونین است، تبدیل می شوند.
آنومالی های تیموس که منجر به تیموس گردنی نابجا می شود ، در اثر سه مکانیسم زیر ممکن است به وجود آید:
1)نزول ناکامل تیموس به قفسه ی سینه
2) باقی ماندن قسمتی از بافت تیموس در مسیر طبیعی نزول تیموس پبه قفسه سینه
3)از بین نرفتن مجرای تیموفارنژیال
آنومالی بعدی کیست های تیموسی است که 2 فرضیه جهت توجیه پاتوژنز آنها وجود دارد:
1)تخریب پیشرونده و اکتسابی باقی مانده های اپیتلیالی یا اجسام هاسال(hassall)
2)بقایای مجرای تیموفارنژیال تشکیل کیست می دهد.
آنومالی پاراتیروئید به صوت ایجاد کیست های پاراتیروئیدی تظاهر میکند.
از لحاظ جنین شناسی این کیست ها یا غدد پاراتیروئید نا بجا می توانند در هر جایی از اطراف عده ی تیروئید پیدا شوند ولی رایج ترین مکان در بخش پایینی غده ی تیروئید است.
به خاطر ارتباط خاستگاهی پاراتیروئید و تیموس، در هرجایی که آنومالی تیموس داشته باشیم امکان وجود آنوملی پاراتیروئید نیز وجود دارد.برای مثال می تواند در مدیاستن تیموس و پاراتیروئید نا بجا پیدا کرد.
کیست های پارا تیروئیدی معمولا از نظر بیوشیمیایی مشکلی ندارند ولی گزارشاتی مبنی بر پاراتیروئیدی ناشی از این کیست ها وجود دارد.
پاتوژنز این کیست ها چند فرضیه را شامل می شود:
1)بقایای جنینی بن بست حلقی سوم و چهارم
2)تخریب آدنومای پاراتیروئید به صورت کیستیک
3)تجمع ترشحات غده وایجاد میکروکیست. به مرور این میکروکیست ها بزرگ شده و کیست را ایجاد می کنند. یا اینکه چند میکروکیست به هم متصل شده و کیست بزرگی را به وجود می آورند.
Scc تیروئید
بسیار نادر ، بسیار کشنده و تماما از سلولهای سنگفرشی تشکیل شده است. از نظر بالینی و آسیبشناسی اشتراکاتی با سرطان آناپلاستیک تیروئید دارد و میتوان آن را به عنوان نوعی از سرطان آناپلاستیک تیروئید در نظر گرفت . مواردی که باید rule out شود :
• متاستاز یا تهاجم مستقیم از سلولهای سنگفرشی اوروفارنکس ، حنجره ، تراشه ،ریه و یا اندامهای دیگر
• سرطان پاپیلاری با کانونهای تمایز سنگفرشی ( در 45-15 % کارسینومهای پاپیلری رخ میدهد )
سایر کارسینومهای تیروئید با تمایز سنگفرشی از جمله کارسینوم موکواپیدرموئید ، کارسینوم اسکلروزان موکواپیدرموئید با ائوزینوفیلی ، CASTLE
مشابه با سرطان آناپلاستیک سالمندان مبتلا به گواتر مزمن را تحتتاثیر قرار میدهد .
تظاهرات بالینی:
بیماران سالمند با رشد سریع توده گردنی مراجعه میکنند – بیماران با سابقهی طولانی بیماری تیروئید – متاستازهای خارج تیروئیدی و به گرههای گردنی متداول است اما متاستازهای دوردست نادر است .
پیشآگهی ضعیف با بقای متوسط 6 ماه – مرگ تقریبا در همه موارد که که معمولا ناشی از پیشرفت لوکال و تحت فشار قرارگرفتن مسیر هوایی است .
درمان : Radical resection و رادیوتراپی
Scc تیروئید
بسیار نادر ، بسیار کشنده و تماما از سلولهای سنگفرشی تشکیل شده است. از نظر بالینی و آسیبشناسی اشتراکاتی با سرطان آناپلاستیک تیروئید دارد و میتوان آن را به عنوان نوعی از سرطان آناپلاستیک تیروئید در نظر گرفت . مواردی که باید rule out شود :
• متاستاز یا تهاجم مستقیم از سلولهای سنگفرشی اوروفارنکس ، حنجره ، تراشه ،ریه و یا اندامهای دیگر
• سرطان پاپیلاری با کانونهای تمایز سنگفرشی ( در 45-15 % کارسینومهای پاپیلری رخ میدهد )
سایر کارسینومهای تیروئید با تمایز سنگفرشی از جمله کارسینوم موکواپیدرموئید ، کارسینوم اسکلروزان موکواپیدرموئید با ائوزینوفیلی ، CASTLE
مشابه با سرطان آناپلاستیک سالمندان مبتلا به گواتر مزمن را تحتتاثیر قرار میدهد .
تظاهرات بالینی:
بیماران سالمند با رشد سریع توده گردنی مراجعه میکنند – بیماران با سابقهی بیماری تیروئید – متاستازهای خارج تیروئیدی و به گرههای گردنی متداول است اما متاستازهای دوردست نادر است .
پیشآگهی ضعیف با بقای متوسط 6 ماه – مرگ تقریبا در همه موارد که که معمولا ناشی از پیشرفت لوکال و تحت فشار قرارگرفتن مسیر هوایی است .
درمان :
Radical resection و رادیوتراپی
تغییرات مولکولی در کارسینوم پاپیلاری تیروئید
اولین واصلی ترین تغییر قابل توجه درPTCبازآرایی کروموزومی RETیا جهش پروتوانکوژن های RASوBRAF است که باعث افزایش فعالیت پروتئین کینازهای میتوژن(MAPK) میشود.که سببافزایش تکثیر سلولی میشوند.
همچنینی مولکول هایی که به صورت فیزیولوژیک باعث رشد تیروسیت ها میشوندمانند اینترلوکین ۱و اینترلوکین۸ و همچنین سایر سایتو کاین ها(مانندIGF1,TGF beta,EGF)میتوانند در ایجاد این نوع از کنسر تیروئید نقشی ایفا نمایند.
افزایش بیان و تولید پروتئین هایی مانندheat shock protein70,,,proxiredoxin,,,isoform s100protein و همچنینی افزایش تولیدmicroRNAها نیز درPTC قابل مشاهده است
Scc تیروئید
بسیار نادر ، بسیار کشنده و تماما از سلولهای سنگفرشی تشکیل شده است. از نظر بالینی و آسیبشناسی اشتراکاتی با سرطان آناپلاستیک تیروئید دارد و میتوان آن را به عنوان نوعی از سرطان آناپلاستیک تیروئید در نظر گرفت . مواردی که باید rule out شود :
متاستاز یا تهاجم مستقیم از سلولهای سنگفرشی اوروفارنکس ، حنجره ، تراشه ،ریه و یا اندامهای دیگر
سرطان پاپیلاری با کانونهای تمایز سنگفرشی ( در 45-15 % کارسینومهای پاپیلری رخ میدهد )
سایر کارسینومهای تیروئید با تمایز سنگفرشی از جمله کارسینوم موکواپیدرموئید ، کارسینوم اسکلروزان موکواپیدرموئید با ائوزینوفیلی ، CASTLE
مشابه با سرطان آناپلاستیک سالمندان مبتلا به گواتر مزمن را تحتتاثیر قرار میدهد .
تظاهرات بالینی:
بیماران سالمند با رشد سریع توده گردنی مراجعه میکنند – بیماران با سابقهی طولانی بیماری تیروئید – متاستازهای خارج تیروئیدی و به گرههای گردنی متداول است اما متاستازهای دوردست نادر است .
پیشآگهی ضعیف با بقای متوسط 6 ماه – مرگ تقریبا در همه موارد که که معمولا ناشی از پیشرفت لوکال و تحت فشار قرارگرفتن مسیر هوایی است .
درمان:
radical resction و رادیوتراپی
پانکراتیت مزمن:
پانکراتیت مزمن معمولاً در پی حملات مکرر پانکراتیت حاد بروز میکند زیرا لوزالمعده در بین حملات کاملاً بهبود نمییابد. لوزالمعده در جریان این عارضه، به تدریج توانایی تولید آنزیمهای گوارشی و هورمونهای ضروری مانند انسولین را از دست میدهد. التهاب پانکراس ناشی از خودهضمی بافت آن توسط آنزیمهای پانکراس به ویژه تریپسین میباشد. علایم پانکراتیت ممکن است به صورت دردهای مداوم یا متناوب در ناحیه فوقانی شکم احساس شوند (علایم با غذاخوردن تشدید میشود و دورههای درد گاه یک هفته طول میکشد). تهوع، استفراغ، اتساع شکم، زردی (یرقان)، اسهال چرب و کاهش وزن.
پانکراتیت اتوایمیون
شیوع این تظاهر از بیماری در ژاپن ٠.٨٢ درصد در هر ۱۰۰ هزار نفر است که به صورت گسترده رو به افزایش است. باید دو فرم پانکراتیت اتوایمیون (AIP) را از هم افتراق دهیم.
اول که مرتبط با اصل نام دیگرش پانکراتیت اسکلروتیک لنفوسیتیک است. فرم دوم که مستقل ازIgG4-RD است، با عناوین AIP-Type2 ,Idiopathic duct centric chronic pancreatitis, و پانکراتیت گرانولوسیتیک اپیتلیالی شناخته می شود.
فرم دوم پانکراتیت نسبت به فرم اول بسیار ناشایع است و گاهی با سندرم روده ملتهب (IBD) همراه است
اکثر بیماران علاوه بر پانکراتیت تیپ ۱ AIP با سایر تظاهرات IgG4-RD همراه هستند مثل کلانژیت اسکلروزتیک، لنفادنوپاتی و یا درگیری غدد بزاقی و لاکریمال.
تشخیص افتراقیAIP1 از آدنوکارسینوم پانکراس بر اساس تظاهرات بالینی برخی اوقات بسیار مشکل است؛ یرقان بدون درد به عنوان مثال یک یافته بسیار شایع در هر دو بیماری است و اکثر بیماران AIP1 بدون در نظر گرفتن سرطان پانکراس، پروسه whipple را متحمل میشوند و همچنین سطح IgG4 افزایش یافته در هر دو بیماری دیده می شود.
یافتههای رادیولوژیک AIP1 شامل بزرگی منتشر پانکراس که به نام پانکراس سوسیسی شکل شناخته میشود و هالهای از ادم گسترده اطراف پانکراس است. هر دوی این ویژگیها در سی تی اسکن شکم به خوبی مشاهده میشود.
سلولهای Tcell-cytotoxic CD4+دارای مولکولی به نام F7( SLAMF7) که یک سیگنال فعال کننده لنفوسیت است، روی سطح خود دارند.
آدنوم هیپوفیز ایزوله خانوادگی (Familial isolated pituitary adenoma) و یا FIPA یک اختلال ارثی است که با رشد یک تومور غیر سرطانی در غده هیپوفیز (آدنوم هیپوفیز-pituitary adenoma) مشخص می شود.
تومورهای ایجاد شده در غده هیپوفیز، می توانند مقدار زیادی از یک یا چند هورمون را آزاد کنند، اگرچه بعضی از این تومورها، هورمون تولید نمی کنند (آدنوم های هیپوفیز غیر عملکردی-nonfunctioning pituitary adenomas). تومورهایی که در تولید هورمون ها اختلال ایجاد می کنند، معمولا از طریق هورمون های خاص تولید شده، تشخیص داده می شوند. پرولاکتینوما ها (Prolactinomas) شایع ترین تومور در FIPA هستند. این تومورها پرولاکتین (prolactin)، هورمون محرک تولید شیر در زنان را تولید می کنند.
علاوه بر این، نوع دیگری از تومور به نام سوماتوتروپینما (somatotropinoma) نیز در FIPA شایع است. این تومورها تولید کننده ی هورمون رشد (که سوماتوتروپین-Somatotropin نیز نامیده می شود)، می باشند که این هورمون باعث رشد بدن میشود. سوماتوتروپینما می تواند در کودکان و یا بزرگسالان منجر به افزایش قد (ژیگانتیسم-gigantism) شود، زیرا رشد استخوان های دراز دست و پا ادامه دارد.
جهش در ژن AIP، می تواند باعث ایجاد FIPA شود. عملکرد پروتئین تولید شده از این ژن به خوبی مشخص نیست اما به نظر می رسد به عنوان یک مهارکننده تومور (tumor suppressor) عمل می کند. مهار کننده های تومور به جلوگیری از رشد و تقسیم غیر قابل کنترل سلول ها، کمک می کنند.
ژنتیک:FIPA دارای الگوی وراثتی اتوزومال غالب است. در این الگوی وراثتی، یک نسخه از ژن تغییر یافته در هر سلول برای ایجاد اختلال کافی است. با این حال، تنها 20 الی 30 درصد افراد دارای جهش در ژن AIP دچار آدنوم هیپوفیز می شوند. این پدیده، که در اثر آن برخی از افراد دارای جهش، دچار علائم بارز یک اختلال نمی شوند، نفوذ پذیری ناقص (incomplete penetrance) نام دارد.