اسلاید آموزشی درباره renal cell carcinoma
Renal Cell Carcinoma from Sam
اسلاید آموزشی درباره بیماری های داخلی کلیه ها
Renal pathology from raj kumar
بافت شناسی گلومرول
اسلاید آموزشی درباره renal cell carcinoma
اسلاید آموزشی درباره بیماری های داخلی کلیه ها
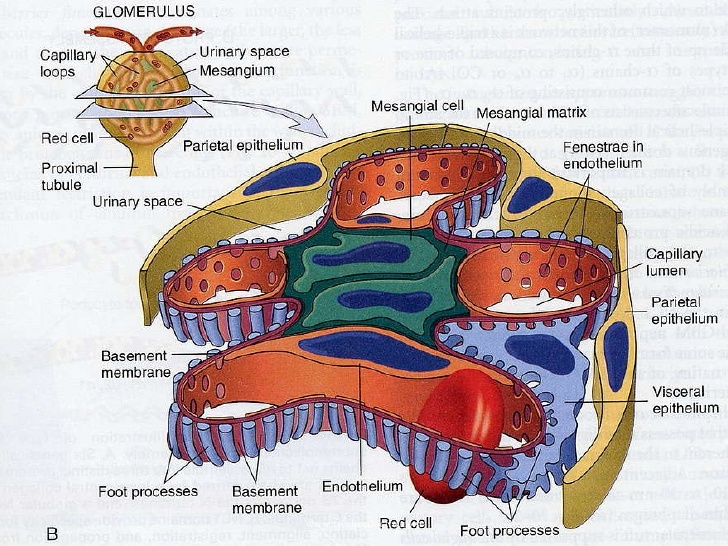
بافت شناسی گلومرول

به ما ملحق شوید..
سندرم آلپورت بيماري ژنتيکي است که در آن ژن يکي از پپتيدهاي سازنده کلاژن نوع IV جهش يافته است و باعث شکننده شدن GBM میشود. اين نوع کلاژن در غشاء پايه گلومرولهاي کليه، گوش داخلي و چشم وجود دارد.
از نظر باليني : علامت برجسته آن هماچوري است كه گاهي خالص و گاهي همراه با آلبومين اوري آمينو اسيداوري و حتي پيوري خواهد بود.کاهش شنوایی و اختلال در بینایی از دیگر علائم این بیماری میباشد.
از نظر پاتولوژی : ضايعات كليوي عبارتند از مخلوطي از ضايعات گلومرولونفريت – پيلونفريت و نفريت اینترسيشيل.
در میکروسکوپ الکترونی GBM ضخیم و نامنظم و تکهتکه میشود و نمای Basket weave مشاهده میشود.
درمان: هیچ درمان شناخته شدهای وجود ندارد و فقط با کنترل فشار خون و محدودیت مصرف پروتئین میتوان از سرعت پیشرفت بیماری کم کرد.پیوند کلیه روش ارجح در موارد نارسائی کلیه است.درمان با ژن درماني و سلول هاي بنيادي هم از جمله روش های درمان می باشد
MPGN: ممبرانو پرولیفراتیو گلومرولونفریت
اتیولوژی:این بیماری در اثر رسوب کمپلکس های ایمنی و یا کمپلمان در مزانژیوم و ساب اندوتلیال ایجاد می شود.به دو دسته تقسیم می شود که در بررسی میکروسکوپیک ایمونوفلورسانس متمایز می شوند.(MPGN با واسطه کمپلکس ایمنی و MPGN با واسطه کمپلمان)
علائم:در برخی بیماران فقط با هماچوری و پروتیینوری غیرنفروتیک تظاهر می کند و در برخی دیگر نمای نفروتیک-نفریتیک مرکب دارد.
تشخیص: با توجه به علائم و یافته های ازمایشگاهی صورت میگیرد.مشخصه بافت شناسی ان تغییر در GBM و مزانژیوم و تکثیر سلول های گلومرولی است.در میکروسکوپ نوری هر دو نوع MPGN مشابه هستند.گلومرول ها بزرگ هستند و یک نمای لوبولی مشخص دارند و تکثیر سلول های مزانژیال و اندوتلیال به همراه لوکوسیت های ارتشاح یافته را نشان می دهد.GBM ضخیم شده و دیواره مویرگ های گلومرولی معمولا حاشیه دو جداره داشته و نمای ریل راه اهن دارند که در رنگ امیزی PAS و نقره مشخص تر است.این حالت در اثر شکافته شدن GBM ایجاد شده است.
درمان:برای درمان ان باید به علت زمینه ای توجه کرد.اما ممکن است این بیماری ایدیوپاتیک باشد که در این صورت درمان ان دشوار است.به طور کلی معمولا اگربیماری به صورت سندروم نفریتیک ظهور کرد از درمان های ایمونوساپرسیو استفاده نمیکنیم اما اگرپروتئینوری ان درحد سندروم نفروتیک بود از استرئیدها استفاده میکنیم (Prednisone)
سیر بیماری:اگر MPGN همراه با یک علت زمینه ای باشد با درمان ان علت میتوان انتظار پروگنوز خوبی را داشت.اما اگر MPGN ایدیوپاتیک باشد نمیتوان پروگنوز خوبی برای بیمار متصور بود(حدود 50-60% افرادی که درمان نمی شوند طی 10-15 سال منجر می شوند به end-stage kidney disease)
(MGN (Membranous Glumerolonephritis:
گلومرونفریت غشایی عامل حدود 30درصد موارد سندرم نفروتیک می باشد و اوج بروز آن در سنین ۳۰ _۵۰ سال بوده ، نسبت ابتلا در مردان به زنان ۲ به ۱ می باشد.در کودکی نادر است اما شایع ترین سندرم نفروتیک در سالمندان محسوب میشود.دونوع دارد ( اولیه ، ثانویه ( که در اثر عفونت ، سرطان ، داروها ، بیماری های خود ایمنی و سیستمیک رخ می دهد ))
اگر چه عوارض ترومبوتیک وجه مشخصه ی تمام سندرم های نفروتیک می باشد ولی MGN بالا ترین میزان بروز گزارش شده ترومبوز وریدی ، امبولی ریوی و ترومبوز ورید عمقی را دارد ، در این افراد ادم ، دیس لیپیدمی و فشار خون بالا مشاهده می شود .
در بررسی بیوپسی کلیه با میکروسکوپ نوری ، ضخیم شدگی یکنواخت غشای پایه در امتداد قوس مویرگ محیطی قابل مشاهده می باشد . ایمونوفلورسانس نشان دهنده ی رسوب منتشر Ig Gو C3است . میکروسکوپ الکترونی به طور تیپیک نشانگر رسوبات الکترون دنس زیر اپی تلیوم است .
استفاده پیشگیرانه از داروهای ضد انعقادی و مهار سیستم رنین انژیوتانسین توصیه میشود . انتخاب داروهای سرکوبگر ایمنی مورد اختلاف نظر است اما در حال حاضر درمان با استروئید ها و سیکلوفسفامید ، کلرامبوسیل ، مایکوفنولات موفتیل ، یا سیکلوسپورین را توصیه می کنند.در بیمارانی که دچار عود بیماری میشوند میتوان ریتوکسیماب ( انتی بادی ضد CD20 )یا استفاده از هورمون ادرنو کورتیکوتروپیک صناعی را در نظر گرفت.
فروکش خود به خودی در ۲۰ تا ۳۳ درصد بیماران دیده می شود . در ۱/۳ بیماران شعله ور شدن سندرم نفروتیک با حفظ کارکرد طبیعی کلیه ادامه می یابد و تقریبا ۱/۳دیگر آن ها به طرف نارسایی کلیه رفته یا در اثر عوارض سندرم نفروتیک جان خود را از دست می دهند.
HCDD یک اختلال رسوب گذاری ایمونوگلوبولین منوکلونال است که ناشی از رسوب زنجیره های سنگین غیر طبیعی ایمونوگلوبولین با ناهنجاری در منطقه CH1 وVH است شواهد نشان میدهد که از دست دادن دامنه CH1 منجر به ترشح زنجیره های سنگین از سلول های پلاسما قبل از اتصال به زنجیره های سبک میشود. همچنین حذف مناطق متغییر زنجیره سنگین (حوزه VH) منجر به تغییر در خواص فیزیکوشیمیایی زنجیره سنگین شده که باعث تغییر هیدروفوبیک و کل بار و در نهایت رسوب بافتی میشود.این رسوبات میتوانند در قلب ،کلیه،کبد،مفاصل وپوست تجمع کنند وباعث نارسایی ارگان مربوطه شوند .
علایم بالینی رایج شامل :سندروم نفروتیک ،نارسایی کلیوی پیشرفته ،فشارخون بالا ،هماچوری .،در بعضی موارد مولتیپل میلوما است .در اغلب بیماران اختلال هماتولوژیک خفیف وبدون میلوم آشکار است.
ازنظر پاتولوژی:میکروسکوپ نوری ، گلومرول اسکلروز ندولار رابا گره های مزانژلی ،افزایش ضخامت دیواره گلومرول و کپسول بومن نشان میدهد .
ودر ایمونوفلورسانس مثبت بودن آن برای آنتی بادی پلی کلونال علیه ساب کلاس igG بدون هیچ گونه مثبت بودن برای زنجیره سبک در TBM.GBM ،کپسول بومن و… است.هم چنین زیر ساخنار میکروسکوپ الکترونی میتواند نشان دهنده رسوبات غلیظ فیبرولار،پودر متراکم درامتداد غشای زیر بغل لوله ای ،غشای زیر بغل گلومرولی و رگ خونی باشد .(HCDD یک بیماری گلومرولی خالص نیست و ضایعات لوله ای به شکل PAS مثبت و ضخیم شدن غشا در نواحی غیر گلومرولی مثل دیستال و هنله مشاهده میشود )
درمان : درمان میتواند به صورت شیمی درمانی با داروهای ملفالان،پردنیزون،تالیدومید،بورتزومیب ،کارموستین یا سایر موارد باشد .
در نهایت اینکه چشم انداز این بیماری خوشبینانه نیست .در صورت درگیر شدن ارگان های اصلی مثل قلب و کبد ، احتمالا طول عمر بیمار بیش از شش ماه نیست .و عفونت معمولا علت اصلی مرگ این بیماران میباشد .
HCDD مخفف Heavy Chain Deposition Disease به معنی بیماری رسوب زنجیره سنگین است .
نفریت لوپوسی:
معرفی بیماری:
در حدود 50 الی 70% بیماران مبتلا به لوپوس شاهد بروز نفریت لوپوسی میباشیم. از این رو در حضور هرگونه پروتئینوری یا سدیمان ادراری فعال یعنی مشاهده ی RBC Cast یا RBC های دیس مورفیک در این بیماران باید بیوپسی کلیوی انجام شود. بر اساس بیوپسی های انجام شده، بیماران مبتلا به نفریت لوپوسی در 6 کلاس مورد طبقه بندی قرار میگیرند.
سیر بیماری:
در کلاس I و II نفریت لوپوسی تنها شاهد تغییرات مزانژیال میباشیم. در کلاس I سدیمان ادراری بیمار نرمال میباشد و در کلاس II علائم بیماران محدود به یک هماچوری و پروتئینوریlow grade می باشد.
در کلاس III به تدریج شاهد درگیری لایه های اصلی گلومرول های کلیوی میباشیم. در این کلاس شاهد سدیمان ادراری فعال و پروتئینوری کمنر از 3gr میباشیم.
کلاس IV این بیماری به صورت نفریت گلوبال یا سگمنتال منتشر میباشد. در این مرحله همانند بیماران مبتلا به MPGN ممکن است شاهد سندرم نفروتیک نفریتیک باشیم.
کلاس V نفریت لوپوسی دارای الگوی ممبرانو بوده و در این بیماران به صورت تیپیک شاهد سندرم نفروتیک میباشیم.
در کلاس VI هم شاهد وقوع اسکلروز پیشرونده در بافت کلیه و شواهد نارسایی مزمن کلیه میباشیم.
*در بین کلاس های فوق کلاس III و IV و VI دارای بدترین پیش آگهی میباشند.
درمان:
کلاس I: پروگنوز در این بیماران عالی است و نیاز به درمان خاصی ندارند.
کلاس II: برای درمان این بیماران بر اساس شدت پروتئینیوری عمل میکنیم:
پروتئینیوری کمتر از 1gr/day : درمان مشابه تظاهرات خارج کلیوی لوپوس
پروتئینیوری بیش از 3gr/day : تجویز کورتون یا داروهای مهار کننده کلسی نورین
کلاس III یا IV : درمان القایی با تجویز کورتون+سیکلوفسفاماید یا مایکوفنولات مافتیل
کلاس V : بیماران واقع در این کلاس معمولا پیش آگهی بسیار خوبی دارند و معمولا فقط تحت درمان حمایتی قرار میگیرند البته در حضور پروتئینیوری پیشرونده یا پایدار در محدوده ی سندرم نفروتیک لازم است به فکر تجویز دارو های زیر به منظور کنترل بیماری باشیم : کورتون + سیکلوسپورین یا مایکوفنولات مافتیل
*در صورت بروز ESRD در این بیماران، بایستی به فکر انجام پیوند کلیه باشیم. خوشبختانه شانس عود این بیماری در کلیه ی پیوندی بسیار پایین میباشد.
بیماری اکتسابی کیستیک کلیه(ACKD):
این بیماری از عوارضCRFاست ودر ۱۰۰—۸۰درصد بیمارانی که به مدت ۱۰سال دیالیز شده اند دیده میشود.
اتیولوژی:علت اصلی،ازبین رفتن آهسته و پیشرونده پارانشیم کلیوی است.با تخریب یکسری نفرون ها،مابقی نفرون های سالم وچار هایپرپلازی میشوند و با ادامه ترشح دراین توبولهای هایپرپلاستیک،کیست ایجاد میشود.
تظاهرات بالینی:وجود بیش از۳تا۵کیست ماکروسکوپی درهرکلیه بیمارانی که فاقد بیماری ارثی کلیوی هستند.
سیر و بروز بیماری:درهمودیالیز و دیالیز صفاقی،بروز یکسانی دارد و نوع دیالیز نقشی در ایجاد کیست ندارد.
سیر بیماری پس از پیوند کلیه متغیر است:
-اگر عملکرد کلیه پیوندی خوب باشد،رشد کیست اهسته یا معکوس میشود
-اگر عملکرد کلیه پیوندی مختل یا پیوند شکست بخورد،احتمال پیشروی در کلیه غیر پیوندی وایجاد احتمالیACKDجدید در کلیه پیوندی وجود دارد.
پیش آگهی:درمبتلایان به ACKDحدود۴۰درصد خطر بروط RCCافزایش میابد که ریسک فاکتورهای آن عبارتند از:جنس مذکر،سیاه پوستان،دیالیز طولانی مدت وACKDشدید همراه با بزرگی شدید کلیه
غربالگری و تشخیص:غربالگری در کلیه پیوندی با سونوگرافی و سپس CTاسکن از ضایعات مشکوک توصیه میشود.
بهترین راه تشخیص CTاست.
درمان:تومور بالای 3cmباید تفروکتومی شود
گلومرولونفریت کرایوگلوبولینمیک:
کرایوگلبولین ها،ایمونوگلوبولین هایی هستند که در دمای پایین رسوب میکنند و با گرم شدن دوباره حل میشوند.
اتیولوژی:علل کرایوگلبولین عبارتنداز :
1-ایدیوپاتیک
2-بیماری های اتوایمیون
3-بدخیمی
4-عفونت:یکی از علل اصلی عفونت با HCV است.
کرایوگلبولین به سه تایپ I, II, III بر اساس نوع ایمونوگلوبولین تقسیم میشود.
تظاهرات بالینی:بیماری کلیوی در 20تا 60 درصد این بیماران رخ می دهد و با پروتئینوری، هماچوری میکروسکوپیک، سندرم نفروتیک و اختلال کلیوی تظاهر پیدا میکند. هایپرتنشن شایع بوده و ممکن است به ویژه در صورت همراهی با سندرم نفزیتیک حلد شدید باشد.
میزان کرایوکریت(cryocrit) با فعالیت بیماری ارتباط ضعیفی دارد.
از منظر پاتولوژیک در میکروسکوپ نوری الگوی ضایعات ممبرانوپرولیفراتیو با واسطه کمپلکس ایمنی مشاهده میشود.
در میکروسکوپ الکترونی، رسوبات منتشر و دنس در ناحیه ساب اپیتلیال به همراه نمای میکروتوبولر یا کریستالی که لوپ های مویرگی را مسدود نموده مشاهده میشود.
درمان:رفع عامل زمینه اساس درمان است. مبتلایان به هپاتيت C فعال باید تحت درمان ضد ویروسی قرار گیرند، مبتلایان به گاموپاتی مونوکلونال باید درمان ضد میلوم دریافت کنند. در کرایوگلبولینمی mixed شدید میتوان از ریتوکسیماب با یا بدون پلاسمافرز استفاده کرد.
پروگنوز کلیوی بیماری خوب بوده و تعداد اندکی از بیماران به سمت ESRD پیشرفت میکنند.
MCD:شایعترین علت سندرم نفروتیک در کودکان (90-70%)و عامل 20-15%موارد سندرم نفروتیک در بالغین است .
علت:اغلب ایدیوپاتیک است .ندرتا ثانویه به بیماری های تیموما و هوچکین یا مصرف NSAIDدیده میشود.
علایم بالینی :ادم ،از دست دادن پروتئین محدود به پروتئین های کوچک(عمدتا آلبومین )،فقدان سلول در ادرار و سایر علایم با شیوع کمتر هایپرتانسیون،هماچوری میکروسکوپی،آتوپی و نارسایی کلیوی است .
تشخیص:در کودکان بر اساس شک بالینی است . در میکروسکوپ نوری به علت تغییرات اندک هیچ یافته ای وجود ندارد توبول ها درجاتی از نفرولیپوز را به صورت سلول های کف آلود حاوی چربی نشان میدهند .در میکروسکوپ ایمنوفلوئورسانس چون Ig نداریم چیزی نمی بینیم اما در میکروسکوپ الکترونی از بین رفتن زوائد پدوسیت دیده میشود .
درمان :اگر چه مشخص شده در 30 % کودکان رمیشن خود به خودی اتفاق می افتد ولی اغلب کودکان با استروئید درمان میشوند.
سیر بیماری: معمولا خود محدود شونده است در کودکان عود پس از اولین رمیشن شایع است و بعد از بلوغ دفعات عود کمتر است .درکل پیش آگهی در کودکان بهتر از بزرگسالان است.
گرانولوماتوز با پلی آنژئیت (GPA) (گرانولوماتوز وگنر)
یک واسکولیت نکروزان است که با تریاد زیر مشخص می گردد:
1-گرانولوم های نکروزان در دستگاه تنفس فوقانی یا دستگاه تنفسی تحتانی یا هر دو.
2-واسکولیت گرانولوماتوز یا نکروزان رگهای با اندازه کوچک تا متوسط با ارجحیت در ریه ها و راههای هوایی فوقانی
3-گلومرولونفریت نکروزان کانونی و اغلب کرسنتیک هلالی شکل
Histopathology
ضایعات راه هوایی فوقانی از سینوزیت گرانولومایی تا ضایعات زخمی بینی، کام یا حلق متفاوت است .یافته های ریوی نیز از ارتشاح پارانشیمی منتشر تا ندول گرانولوماتوز متفاوت می باشد.
تخریب رگها می تواند منجر به خونریزی و هموپتيزی گردد. ضایعات نهایتا ممکن است دچار فیبروز پیشرونده و سازمان یابی شوند.
ضایعات کلیوی از یک نکروز کانونی خفیف در گلومرول همراه با ترومبوز مجرای لوپ های مویرگی گلومرولی (گلومرولونفریت نکروزان کانونی و سگمنتال) تا ضایعات گلومرولی پیشرفته تر با نکروز منتشر و تکثیر سلولی پاریتال همراه با تشکیل هلال های اپی تلیالی ( گلومرولونفریت هلالی) متغیر هستند.
ویژگی های بالینی
اشکال محدود بیماری می تواند محدود به دستگاه تنفس باشد. بالعکس، شکل بیماری گسترده می تواند چشمها، پوست و سایر ارگان ها به ویژه قلب را درگیر کند؛ GPA منتشر از نظر بالینی، شبیه پلی آرتریت ندوزا به علاوه درگیری ریوی می باشد. گرانولوماتوز با پلی آنژئیت (GPA) احتمالا به صورت پاسخ افزایش حساسیت با واسطه سلول، علیه آنتی ژن های عفونی یا محیطی استنشاق شده آغاز می شود.
– سطح ANCA نشانگر مفیدی برای فعالیت بیماری است.
بیمار معمولا یک مرد میانسال است، گرچه زنان و گروههای سنی دیگر نیز می توانند مبتلا شوند. تظاهرات کلاسیک، شامل پنومونیت دوطرفه به همراه ضایعات حفرهای و ندولار (۹۵%)، سینوزیت مزمن (۹۰٪)، زخمهای مخاطی نازوفارنکس (۷۵%) و بیماری کلیوی (۸۰٪) می باشد. بیماران مبتلا به درگیری کلیوی خفيف ممکن است تنها هماچوری و پروتئینوری را نشان دهند .
. در صورت عدم درمان، ۸۰٪ بیماران در عرض ۱ سال از بین می روند.
درمان
از : استروئیدها، سیکلوفسفامید، مهارکننده های TNF و ریتوکسیماب برای درمان وگنر استفاده میشود.
پلی ارتریت ندوزا (PAN) یک واسکولیت عروق متوسط است که با انوریسم های شریانی و تنگی شریان های عضلانی مشخص می شود. تنگی ها به صورت سگمنتال و در مناطق دو شاخه شدن شریان ها رخ می دهد.دلیل ایجاد این بیماری هنوز یافت نشده است ولی این بیماری به صورت اولیه یا ثانویه به عفونت های هپاتیت B و C و HIV دیده شود.شایع ترین تظاهرات بالینی مربوط به دستگاه گوارش، کلیه و سیستم عصبی می باشد(انژین روده ای،هیپرتنشن، منونوریت مولتی پلکس). تشخیص این بیماری بر اساس یافته های انژیوگرافی یا بیوپسی و بررسی عفونت های هپاتیت B و C و HIV است.درمان پلی ارتریت ندوزا شامل کورتیکواستروئید ها یا دارو های NSAID یا هر دو است و اگر بیماری شدید باشد از دارو های سرکوبگر سیستم ایمنی مانند سیکلوفسفامید استفاده می کنیم.
مراحل (staging ) نفریت لوپوسی
این سیستم طبقه بندی در سال ۲۰۰۳ توسط انجمن بین المللی نفرولوژی و پاتولوژی کلیه پایه گذاری شده است.
classI:Minimal mesangial glomerulonephritis
از نظر بافت شناسی در میکروسکوپ نوری ظاهر نرمال دارند ولی رسوب مزانژیال را میتوان در میکروسکوپ الکترونی مشاهده کرد. این بیماران پیش آگهی عالی داشته و نیاز به درمان ندارند.
class II : Mesangial proliferative glomerulonephritis
مشخصه آن افزایش سلولهای مزانژیال و گسترش ماتریکس می باشد.در این افراد هماچوری میکروسکوپی دیده میشود و تظاهرات آن میتواند با یا بدون پروتئینوری باشد. در این مرحله هایپرتنشن، سندروم نفروتیک و CKD بسیار نادر است.
classIII:Focal proliferative nephritis
این بیماری با ضایعات اسکلروتیکی همراه است که کمتر از ۵۰ درصد گلومرول ها را درگیر میکند.
از نظر بافت شناسی در زیر میکروسکوپ الکترونی رسوب ساب اندوتلیال دیده می شود(گاهی به همراه تغییراتی در سلولهای مزانژیال). ازنظر ایمونوفلورسانس lgA,lgG,lgM و C3 مثبت میشود.
از نظر بالینی هماچوری و پروتئینوری داریم. همچنین هایپرتنشن، سندروم نفروتیک و کراتینین بالای سرم ممکن است دیده شوند.
classIV :Diffuse proliferative nephritis
شدید ترین و شایع ترین فرم نفریت لوپوسی می باشد که در آن بیش از ۵۰ درصد گلومرول ها درگیر می باشند. ضایعات می توانند کلی یا سگمنتال و حاد یا مزمن باشد. رسوب ساب اندوتلیال و تغییرات مزانژیال در زیر میکروسکوپ الکترونی دیده می شود از نظر بالینی با هماچوری،پروتئینوری،کراتینین بالا و اغلب اوقات سندروم نفروتیک و هایپر تنشن بروز پیدا میکند.
و همچنین hyopcopelementemia و تیتر بالای dsDNA از ویژگی های این مرحله می باشد.
Class V:Membranous glomerulonephritis
این مرحله با نازک شدن غشا و تخریب دیواره مویرگ ها مشخص می گردد.
از نظر بالینی نشانه های سندروم نفروتیک دیده می شود. همچنین در این مرحله عوارض ترومبوتیک نظیر ترومبوز عروق رنال یا آمبولی ریوی شایع می باشد.
Class VI:advanced sclerosing lupus nephritis.
در این مرحله ضایعات منتشر اسکلروتیک بیش از ۹۰ درصد گلومرول ها را درگیر میکنند. و معمولاً نشان دهنده یک آسیب التهابی قبلی می باشد.
مشخصه اصلی این مرحله نارسایی پیشرونده کلیه می باشد.
گلومرولونفریت کرایوگلوبولینمیک:
کرایوگلبولین ها،ایمونوگلوبولین هایی هستند که در دمای پایین رسوب میکنند و با گرم شدن دوباره حل میشوند.
اتیولوژی:علل کرایوگلبولین عبارتنداز :
1-ایدیوپاتیک
2-بیماری های اتوایمیون
3-بدخیمی
4-عفونت:یکی از علل اصلی عفونت با HCV است.
کرایوگلبولین به سه تایپ I, II, III بر اساس نوع ایمونوگلوبولین تقسیم میشود.
تظاهرات بالینی:بیماری کلیوی در 20تا 60 درصد این بیماران رخ می دهد و با پروتئینوری، هماچوری میکروسکوپیک، سندرم نفروتیک و اختلال کلیوی تظاهر پیدا میکند. هایپرتنشن شایع بوده و ممکن است به ویژه در صورت همراهی با سندرم نفزیتیک حلد شدید باشد.
میزان کرایوکریت(cryocrit) با فعالیت بیماری ارتباط ضعیفی دارد.
از منظر پاتولوژیک در میکروسکوپ نوری الگوی ضایعات ممبرانوپرولیفراتیو با واسطه کمپلکس ایمنی مشاهده میشود.
در میکروسکوپ الکترونی، رسوبات منتشر و دنس در ناحیه ساب اپیتلیال به همراه نمای میکروتوبولر یا کریستالی که لوپ های مویرگی را مسدود نموده مشاهده میشود.
درمان:رفع عامل زمینه اساس درمان است. مبتلایان به هپاتيت C فعال باید تحت درمان ضد ویروسی قرار گیرند، مبتلایان به گاموپاتی مونوکلونال باید درمان ضد میلوم دریافت کنند. در کرایوگلبولینمی mixed شدید میتوان از ریتوکسیماب با یا بدون پلاسمافرز استفاده کرد.
پروگنوز کلیوی بیماری خوب بوده و تعداد اندکی از بیماران به سمت ESRD پیشرفت میکنند.
C1Qنفروپاتی یک پروتئینوری یا سندرم نفروتیک اسیمپتوماتیک مقاوم به استروئید است و بیشتر در بچه ها و سیاه پوستان و زنان دیده میشود در سنین جوانی و بسیار شبیه به FSGS یا MCDمیباشد..این بیماری که به دلیل رسوب پروتئینC1Qکه یکی از اجزای کمپلکس ایمنی است در بافت مزانژیال کلیه بوجود میاید یا در ادرار دیده میشود و علت این پدیده شناخته نشده است.علائم ان شامل ادم و ورم کردن خصوصا در اندام تحتانی و کمربند لگنی است و افزایش فشار خون و هایپر کلسترولمیا و مستعد شدن به تشکیل لخته با یا بدون هماچوری گروس.تشخیص ان با بیوپسی کلیه و میکروسکوپ ایمونوفلورسانس:دیدن زنجیره های C1Qدامیننت یا کودامیننت و نمایFULLHOUSE WITH DEPOSITES OF IGG ,IGM , IGA,C1Q,C3ومیکروسکوپ الکترونی با دیدن امورف های الکترون دنس.درمان شامل داروهای ACEI,ARBSوسرکوب کننده های ایمنی مثل سیکلوسپورین و ازاتیوپرین است.این بیماری ممکن است به FSGSیا RENALFAILUREیاCKDبیانجامد.
پیوند کلیه باعث افزایش کیفیت زندگی و بقای بیماران ESRD شده است. رد پیوند آلوگراف کلیه به دو دسته وابسته به Tسل (TCMR) و وابسته به آنتی بادی (ABMR) تقسیم می شود که هر دو از نظر یافته های پاتولوژی با طبقه بندی Banff قابل تشخیص اند.
۱- Acute ABMR: معمولاً ۱ تا ۳ هفته بعد از پیوند رخ می دهد. التهاب عروق ریز و اتساع عروق اطراف توبولی به همراه حضور نوتروفیل ها مشاهده می شود. آرتریت اینتیمال یا ترنس مورال هم یافت می شود.
۲- Chronic ABMR: به دنبال تولید آنتی بادی DSA و واکنش آن با اندوتلیوم عروق ریز رخ می دهد که با مثبت شدن C4d همراه است. تغییرات اختصاصی مثل چند لایه شدن غشای پایه (هر لایه نمایانگر یک دوره آسیب) به همراه التهاب، فیبروز لایه اینتیمای شریانی و افزایش کراتینین دیده می شود.
درمان در ABMR بر پایه کاهش سریع تیتر آنتی بادی با پلاسمافرز، ایمونوگلوبین داخل وریدی و ریتوکسیماب و در TCMR بر استروئیدتراپی استوار است.
۳- Acute TCMR: با التهاب توبولی و ارتشاح لنفوسیت های T و ماکروفاژها در بافت بینابینی مشخص می شود.
۴- Chronic TCMR: بر اساس طبقه بندی Bnaff با آرتریوپاتی همراه با اسکلروز مشخص می شود. این پیدده به علت تجمع کلاژن های نوع I و II، فقدان الاستیسیتی و درجات متغیری از التهاب رخ می دهد.
بیماری ضد غشای پایه گلومرولی
بیمارانی که اتوآنتی بادی هایی علیه آنتی ژن های غشای پایه ی گلومرولی تولید می کنند غالبا دچار گلومرونفریتی به نام بیماری ضدغشای پایه گلومرولی ( آنتی GBM) می شوند.وقتی که علائم خونریزی ریوی و گلومرونفریت نشان دهند, یک سندروم ریوی _کلیوی به نام سندروم گودپاسچرنامیده می شود.
RPGM وخونریزی ریوی تظاهر اصلی این سندروم هستند.درافرادمشکوک به سندروم گودپاسچر بیوپسی فوری کلیه مهم است به طورتیپیک بیوپسی کلیه نشان دهنده نکروزفوکال یا سگمنتال است که بعدها همراه با تخریب پیش رونده مویرگ توسط تکثیر سلولی,منجربه تشکیل هلال درفضای بومن می شود.در رنگ آمیزی ایمنوفلورسانس, آنتی بادی علیه زنجیره آلفا 3 کلاژن نوع 4(COLA4A3) به صورت خطی درامتداد GBMو غشای پایه آلوئولی مشاهده می گردد.
ابتداپالس متیل پردنیزولونHigh dose وسپس پردنیزولون با دوز 1mg/kgبه همراه سیکلوفسامید خوراکی تجویزمی گردد.تعویض پلاسما درمان دیگر این بیماران است.
پیش آگهی به درصد کرسنت های محیطی در بیوپسی کلیه,اولیگوری ونیاز به دیالیز بستگی دارد.فاکشن کلیه دربیمارانی که کرسنت ها(هلال ها) صد درصد گلومرول را اشغال کرده اند یا تحت دیالیز قرار دارند بهبود پیدا نمی کنند.
آرتریت تاکایاسو یک بیماری التهابی و انسدادی شریان های متوسط تا بزرگ است.تمایل بیشتری به قوس آئورت و شاخه های آن دارد.این بیماری یک پان آرتریت با اینفیلتراسیون سلول تک هسته ای التهابی و گاها سلول های غول آسا می باشد.پرولیفراسیون و فیبروز شدید اینتیما ، اسکار و واسکولاریزاسیون در مدیا و پارگی و دژنراسیون لامینای الاستیک وجود دارد.باریک شدگی لومن با یا بدون ترومبوز اتفاق می افتد.
علائم عمومی عبارتند از: کسالت،تب،تعریق شبانه،ارترالژی،بی اشتهایی،کاهش وزن. درگیری شریان های کلیه در قالب هایپرتنشن و نارسایی کلیه خود را نشان می دهد.
یافته های آزمایشگاهی عبارتند از : افزایش ESR، آنمی خفیف، افزایش سطح ایمونوگلوبولین
تشخیص : خانم جوانی با کاهش یا فقدان نبض های محیطی، اختلاف فشار خون ، بروئی شریان باید به شدت مورد شک تاکایاسو قرار بگیرد.
الگوی تشخیص آرتریوگرافی:دیوراه عروقی نامنظم،تنگی، اتساع پس از تنگی، تشکیل آنوریسم، انسداد.
نشان دادن هیستوپاتولوژیک عروق ملتهب موجب تایید اطلاعات می شود با این حال به ندرت بافت برای آزمایش در دسترس است.
درمان : گلوکوکورتیکوئید ها برای علایم حاد
جراحی تهاجمی یا آرتریوپلاستیک برای عروق تنگ شده،تصحیح هایپرتنشن ناشی از تنگی شریان کلیوی، افزایش فشار خون احشا و اندام های ایسکمیک
میزان مرگ و میر 5ساله بین 0-35%است . به دلایل: نازسایی احتقانی قلب،حوادث عروقی مغزی،انفارکتوس میوکاردپارگی آنوریسم،نارسایی کلیوی
گلومرولونفريت هلالى ايديوپاتيک به انواع زیر تقسیم می شود؛ ۱. تيپ I- با رسوب خطى Ig (بهواسطه آنتىبادى ضدGBM)۲. تيپ II- با رسوب گرانولر Ig (بهواسطه کمپلکس ايمني)۳. تيپ III- با رسوب کم Ig يا بدون رسوب (”پاسى ايميون“)(Pauci-Immune)
۴. ناشى از آنتىبادى ضدٌ نوتروفيل سيتوپلاسميک
**پاسی ایمیون گلومرونفریت با علائم کلیوی RPGN (هماچوری ، فشار خون بالا) منجر به نارسایی کلیه می شود ، و ممکن است با تظاهرات واسکولیت سیستماتیک (آرتریالژی ، تب ، تشنج و درگیری ریه ها) همراه باشد. ژن مهمی که با گلومرولونفریت Pauci-Immune مرتبط است BMP7 می باشد.طبقه بندی ایمونولوژیک بر اساس وجود یا عدم وجود آنتی بادی های سیتوپلاسمی در گردش ضد نوتروفیل (ANCAs) به دو دسته ی PIGN همراه با ANCA(در ۸۰تا ۹۰ درصد موارد) و بدون آن تقسیم می شود.در بیوپسی کلیوی از ویژگیهای بارز بیماری هلال و نکروز سگمنتال بر میکروسکوپ نوری و عدم وجود رسوبات ایمنی در میکروسکوپ ایمونوفلورسانس را می توان نام برد.درمان با کورتیکواستروئیدهای با دوز بالا به علاوه سرکوب کننده های سیستم ایمنی بوسیله سیکلوفسفامید یا آزاتیوپرین توصیه می شود.
PSGNپست استرپتوکوکال گلومرونفریت
شکل کلاسیک یکacute GNاست که ۱-۴هفته بعد از یک فارنژیت و یا زرد زخم پوستی در اثر استرپتوکوک بتاهمولیتیک گروه Aایجاد میشود و شروع ناگهانی یک سندروم نفروتیک شامل فشار خون بالا سردرد ادرار تیره درد پهلو است برخی علائم پایدار ان شامل هماچوری فشارخون بالا اختلال عملکرد کلیوی است
در تشخیص ان اغلب تمام کشت ها منفی است تیتر انتی بادی Aso_انتی هیالورونیداز،anti_DNAseBبالاست.سطح C3پایین و C4پایین یا نرمال است در Ifرسوب igG+c3و بندرتigm دیده میشود .
درمان استفاده از دیورتیک کنترل فشارخون ریشه کن کردن عفونت فعال است در کودکان پیش اگهی عالی است و در طی دوماه تمام علائم برطرف میشود ولی بندرت در برخی بیماران ممکن است ب مدت طولانی علائم باقی بماند
نفرو پاتی دیابتی به بیماری کلیوی ثانویه به بیماری دیابت گفته میشود.در نفروپاتی دیابتی افزایش پیشرونده دفع پروتئین از ادرار دیده میشودو منجر به افت کارکرد کلیه و در نهایت نارسایی کلیه میشود.دفع آلبومین در بیماران مبتلا به دیابت در ابتدا در حد طبیعی است در مراحل اولیه بیماری دفع آلبومین در ادرار اندک است .میزان دفع اندک اندک افزایش می یابد تا در طول پیشرفت نفروپاتی به دفع واضح پروتئین در ادرار می انجامد. علائم این بیماری شامل:1.افزایش دفع آلبومین و پروتئین از ادرار2.فشار خون بالا3.کاهش فعالیت کلیه ها4.از دست دادن یا افزایش وزن غیر ارادی وکاهش اشتها5.تهوع و استفراغ6.پرنوشی و تشنگی بیش از حدوپرادراری میباشدسیر بیمای:نفروپاتی دیابتی مراحل گوناگون دارد که برای غربالگری وتشخیص و تعیین پیش آگهی بیماری بکار میرود.مراحل نفروپاتی دیابتی شامل:1.هیپرفیلتراسیون گلومرولی و بزرگ شدن کلیه ها2.آسیب های اولیه گلومرولی3.میکروآلبومینوری4.ماکروآلبومینوری وکاهش GFRودر نهایت5.نارسایی کلیه میباشد. تشخیص وتعیین پیشرفت بیماری بر اساس اندازه گیری آلبومین ادرار میباشد همچنین در مرحله آسیب اولیه گلومرولی ضخیم شدن غشاپایه و افزایش ماتریکس مزانژیوم دیده میشود.
نفریت لوپوسی:
معرفی بیماری:
در حدود 50 الی 70% بیماران مبتلا به لوپوس شاهد بروز نفریت لوپوسی میباشیم. از این رو در حضور هرگونه پروتئینوری یا سدیمان ادراری فعال یعنی مشاهده ی RBC Cast یا RBC های دیس مورفیک در این بیماران باید بیوپسی کلیوی انجام شود. بر اساس بیوپسی های انجام شده، بیماران مبتلا به نفریت لوپوسی در 6 کلاس مورد طبقه بندی قرار میگیرند.
سیر بیماری:
در کلاس I و II نفریت لوپوسی تنها شاهد تغییرات مزانژیال میباشیم. در کلاس I سدیمان ادراری بیمار نرمال میباشد و در کلاس II علائم بیماران محدود به یک هماچوری و پروتئینوریlow grade می باشد.
در کلاس III به تدریج شاهد درگیری لایه های اصلی گلومرول های کلیوی میباشیم. در این کلاس شاهد سدیمان ادراری فعال و پروتئینوری کمتر از 3gr میباشیم.
کلاس IV این بیماری به صورت نفریت گلوبال یا سگمنتال منتشر میباشد. در این مرحله همانند بیماران مبتلا به MPGN ممکن است شاهد سندرم نفروتیک نفریتیک باشیم.
کلاس V نفریت لوپوسی دارای الگوی ممبرانو بوده و در این بیماران به صورت تیپیک شاهد سندرم نفروتیک میباشیم.
در کلاس VI هم شاهد وقوع اسکلروز پیشرونده در بافت کلیه و شواهد نارسایی مزمن کلیه میباشیم.
*در بین کلاس های فوق کلاس III و IV و VI دارای بدترین پیش آگهی میباشند.
درمان:
کلاس I: پروگنوز در این بیماران عالی است و نیاز به درمان خاصی ندارند.
کلاس II: برای درمان این بیماران بر اساس شدت پروتئینیوری عمل میکنیم:
پروتئینیوری کمتر از 1gr/day : درمان مشابه تظاهرات خارج کلیوی لوپوس
پروتئینیوری بیش از 3gr/day : تجویز کورتون یا داروهای مهار کننده کلسی نورین
کلاس III یا IV : درمان القایی با تجویز کورتون+سیکلوفسفاماید یا مایکوفنولات مافتیل
کلاس V : بیماران واقع در این کلاس معمولا پیش آگهی بسیار خوبی دارند و معمولا فقط تحت درمان حمایتی قرار میگیرند البته در حضور پروتئینیوری پیشرونده یا پایدار در محدوده ی سندرم نفروتیک لازم است به فکر تجویز دارو های زیر به منظور کنترل بیماری باشیم : کورتون + سیکلوسپورین یا مایکوفنولات مافتیل
*در صورت بروز ESRD در این بیماران، بایستی به فکر انجام پیوند کلیه باشیم. خوشبختانه شانس عود این بیماری در کلیه ی پیوندی بسیار پایین میباشد.
بیماری مدولاری کیستیک کلیه (MCKD) توارث اتوزومال غالب دارد و فقط حاشیه کورتیکو مدولاری را درگیر میکند و در اثر آتروفی توبولهای کلیه کوچک میشوند. این بیماران در دهه های 4 تا 6 زندگی مستعد سنگ کلیه هستند.
این بیماری دو نوع دارد: نوع1- جهش در ژن موسین1 (MUC1)
نوع2- جهش در ژن یورومودولین (UMOD)
علایم: افزایش در اسید اوریک خون و کمی پروتئینوری و گاه کیست در سونوگرافی دیده میشود.
بیوپسی کلیه: فیبروز اینترسشیال و آتروفی توبولی دیده میشود و نمای تشخیصی مهم آن نمای قلموی نقاشی مربوط به توبول دیستال است.
درمان: با داروهای کاهنده اسید اوریک مثل آلوپورینول یا فبوکسوستات درمان میشوند.
سندروم نفروتیک مادرزادی فنلاندی :
یک اختلال کلیوی است که زمان آغاز علائم این اختلال در کودکان بین تولد تا سه ماهگی است و تا اوایل کودکی منجر به نارسایی برگشت ناپذیر کلیه میشود.
در اکثر موارد در اکثر موارد در اثر جهش در ژن های NPHS1و NPHS2 ایجاد میشود که باعث از بین رفتن پروتئین کاربردی که در سطح پودوسیت ها و در ناحیه ی Slit diaphragms قرار دارد میشود و باعث ایجاد اختلال در این ناحیه میشود و بدون وجود این ناحیه مولکول های بیشتری (پروتئین ها و گلبول های قرمز) به طور غیرطبیعی از کلیه ها عبور کرده و از ادرار دفع می شوند و در نهایت میتواند منجر به بیماری end stage renal شود.
جهش در ژن NPHS1 باعث ایجاد تمام موارد تیپ فنلاندی سندروم نفروتیک مادرزادی میشود.
علائم:پروتئینوری، هیپر کلسترولمی، آسیت، ادم، هماچوری، آنمی، ایجاد غیرطبیعی لخته و کاهش گلبول های سفید که در نهایت منجربه تضعیف سیستم ایمنی و عفونت های مکرر میشود.
تشخیص:اصلی ترین آزمایش در بیماران دارای سندروم نفروتیک اندازه گیری میزان پروتئین 24 ساعته ی ادرار است، آزمایش خون، بیوپسی و نمونه برداری از کلیه، اکوگرافی و سرم الکتروفورز نیز برای تشخیص استفاده میشود.
علیرضا محمدی
فیزیوپات 1
توضیح:عفونت با E.coli مولد شیگاتوکسین (سوش O157:H7) در ۱۵ درصد موارد با میکروانژیوپاتی ترومبوتیک محدود به عروق کلیوی همراه است که به HUSمرسوم می باشد. HUS به صورت کم خونی همراه با وجود شیستوسیت ها، ترومبوسیتوپنی و اختلال عملکرد کلیه در غیاب دیگر علل اختلال انعقادی تعریف میشود.HUS بیشتر به حالت خوشه ای رخ میدهد که حداکثر شیوع آن در تابستان و پاییز است و در موارد ابتلا، اغلب می توان شواهدی از مصرف گوشت کم پخته شده، تماس با مدفوع گاو، تماس با حیوانات یا دیگر فرآورده های غذایی آلوده را یافت.
علائم بالینی: شیگاتوکسین معمولا علائم پرودرمال اسهال دردناک و خونی ایجاد می کند که 2تا12 روز (متوسط ۳ روز )پیش از HUS رخ می دهد. سم شیگا به طور مستقیم در عروق کلیه ترومبوز ایجاد میکند.
یافته های آزمایشگاهی : افزایش کراتینین، کم خونی، شیستوسیتوز در اسمیر خون محیطی، افزایش شمار رتیکولوسیت ها و ترومبوسیتوپنی.
تشخیص: باید نمونه مدفوع تازه برای کشت E.coli O157:H7ارسال شود.( بررسی مدفوع باید در بیماران فاقد اسهال نیز انجام شود زیرا این پاتوژن ممکن است بندرت در غیاب علائم روده ای،HUS ایجاد کند).
یافته های پاتولوژی: ضخامت دیواره عروق همراه با تورم سلول های اندوتلیال و ترومبوز درون گلومرول همراه با ترومبوز های غنی از پلاکت و فیبرین.
درمان : درمان حمایتی است و شامل جایگزینی کافی مایعات ایزوتونیک، انتقال خون( در موارد کم خونی شدید) است.ترانسفوزیون پلاکت توصیه نمی شود چون ممکن است ترومبوز را در عروق کوچک تشدید کند.درمان آنتی بیوتیکی مبتلایان به اسهال خونی توصیه نمی شود زیرا این درمان در کاهش میزان بروز HUS موثر نیست و ممکن است گاه خطر ابتلا به آن را نیز افزایش دهد .همچنین بی تاثیری کورتیکواستروئیدها، داروهای ضد انعقادی (آسپرین،هپارین)داروهای ضد ترومبوز و پلاسمافرز نیز در درمان HUSاثبات شده است.
(MGN staging (Membranous Glumerolonephritis :
گلومرونفریت غشایی به علت رسوب ایمونوگلوبولین ها در GBM رخ میدهد. که به دو دسته تقسیم میشود:
1) گلومرونفریت غشایی اولیه(primary) : در 85% موارد شاهد این نوع از گلومرونفریت هستیم که به علت ایدیوپاتیک رخ میدهد.در این حالت به علت وجود بیماری خودایمنی که آنتی بادی هایی که با اتوآنتی ژن های ناشناخته واکنش میدهند، رخ میدهد.
2)گلومرونفریت غشایی ثانویه(secondary) :علت به وجود آمدن آن،وجود یک بیماری زمینه ای میباشد از قبیل: الف) عفونت ها ( هپاتیت B مزمن، سیفلیس، شیستومیازیس، مالاریا) ب) تومورهای بدخیم(کارسینومای ریه و کولون ،ملانوما) پ) لوپوس و سایر بیماری های خود ایمنی ت) تماس با برخی فلزات مانند طلا ث) داروها( کپتوپریل، NSAIDs ) .
نمونه ی آزمایشگاهی برای گلومرونفریت غشایی، Heymann نام دارد که توسط renal tubular brush border proteins در حیوانات القا میشود که نهایتا آنتی بادی هایی که تولید میشوند با آنتی ژن های موجود در GBM واکنش میدهند و باعث رسوب گرانولار و پروتئینوری البته بدون التهاب شدید میشوند.
** گرانولوماتوز با پلی آنژئیت (GPA) (گرانولوماتوز وگنر)
یک واسکولیت نکروزان است که با تریاد زیر مشخص می گردد:
1- گرانولوم های نکروزان در دستگاه تنفس فوقانی یا دستگاه تنفسی تحتانی یا هر دو.
2- واسکولیت گرانولوماتوز یا نکروزان رگهای با اندازه کوچک تا متوسط با ارجحیت در ریه ها و راههای هوایی فوقانی
3- گلومرولونفریت نکروزان کانونی و اغلب کرسنتیک هلالی شکل
* Histopathology
ضایعات راه هوایی فوقانی از سینوزیت گرانولومایی تا ضایعات زخمی بینی، کام یا حلق متفاوت است .یافته های ریوی نیز از ارتشاح پارانشیمی منتشر تا ندول گرانولوماتوز متفاوت می باشد.
تخریب رگها می تواند منجر به خونریزی و هموپتيزی گردد. ضایعات نهایتا ممکن است دچار فیبروز پیشرونده و سازمان یابی شوند.
ضایعات کلیوی از یک نکروز کانونی خفیف در گلومرول همراه با ترومبوز مجرای لوپ های مویرگی گلومرولی (گلومرولونفریت نکروزان کانونی و سگمنتال) تا ضایعات گلومرولی پیشرفته تر با نکروز منتشر و تکثیر سلولی پاریتال همراه با تشکیل هلال های اپی تلیالی ( گلومرولونفریت هلالی) متغیر هستند.
* ویژگی های بالینی
اشکال محدود بیماری می تواند محدود به دستگاه تنفس باشد. بالعکس، شکل بیماری گسترده می تواند چشمها، پوست و سایر ارگان ها به ویژه قلب را درگیر کند؛ GPA منتشر از نظر بالینی، شبیه پلی آرتریت ندوزا به علاوه درگیری ریوی می باشد. گرانولوماتوز با پلی آنژئیت (GPA) احتمالا به صورت پاسخ افزایش حساسیت با واسطه سلول، علیه آنتی ژن های عفونی یا محیطی استنشاق شده آغاز می شود.
– ANCA در تقریبا ٪۹۵ موارد مثبت شده و احتمالا آسیب بافتی را باعث می شود.
بیمار معمولا یک مرد میانسال است، گرچه زنان و گروههای سنی دیگر نیز می توانند مبتلا شوند. تظاهرات کلاسیک، شامل پنومونیت دوطرفه به همراه ضایعات حفرهای و ندولار (۹۵%)، سینوزیت مزمن (۹۰٪)، زخمهای مخاطی نازوفارنکس (۷۵%) و بیماری کلیوی (۸۰٪) می باشد.
. در صورت عدم درمان، ۸۰٪ بیماران در عرض ۱ سال از بین می روند.
* درمان
از : استروئیدها، سیکلوفسفامید، مهارکننده های TNF و ریتوکسیماب برای درمان وگنر استفاده میشود.
پلی ارتریت ندوزا (PAN) یک واسکولیت عروق متوسط است که با انوریسم های شریانی و تنگی شریان های عضلانی مشخص می شود. تنگی ها به صورت سگمنتال و در مناطق دو شاخه شدن شریان ها رخ می دهد.برخلاف واسکولیت عروق کوچک درگیری کلیوی در پلی ارتریت ندوزا گلومرولونفریت نبوده بلکه موجب انوریسم و تنگی شریان کلیوی می شود.دلیل ایجاد این بیماری هنوز یافت نشده است ولی این بیماری به صورت اولیه یا ثانویه به عفونت های هپاتیت B و C و HIV دیده شود.شایع ترین تظاهرات بالینی مربوط به دستگاه گوارش، کلیه و سیستم عصبی می باشد(انژین روده ای،هیپرتنشن، منونوریت مولتی پلکس). تشخیص این بیماری بر اساس یافته های انژیوگرافی یا بیوپسی و بررسی عفونت های هپاتیت B و C و HIV است.درمان پلی ارتریت ندوزا شامل کورتیکواستروئید ها یا دارو های NSAID یا هر دو است و اگر بیماری شدید باشد از دارو های سرکوبگر سیستم ایمنی مانند سیکلوفسفامید استفاده می کنیم.
گلومرولونفریت کرایوگلوبولینمیک:کرایوگلوبولین ها ایمنوگلوبولین هایی هستند که در دمای پایین رسوب می کنند و با گرم شدن دوباره حل می شوند.
اتیولوژی:علل کرایوگلوبولینمی عبارتند از:1- ایدیوپاتیک 2-بیماری های اتوایمیون 3-بدخیمی ها 4- عفونت:عفونت با HVC یکی از علل اصلی کرایوگلوبولینمی است.
این بیماری به سه تایپ بر اساس نوع ایمنوگلوبولین تقسیم میشود.
تظاهرات بیماری:بیماری کلیوی در 20-60% این بیماران رخ می دهد و با پروتئینوری ، هماچوری میکروسکوپیک ،سندرم نفروتیک و اختلال کلیوی تظاهر پیدا میکند.هایپرتنشن شایع بوده و ممکن است به ویژه در صورت همراهی با سندرم نفریتیک حاد،شدید باشد..
از منظر پاتولوژی در میکروسکوپ نوری الگوی ضایعات ممبرانوپرولیفراتیو با واسطه ی کمپلکس ایمنی مشاهده می شود.
در میکروسکوپ الکترونی رسوبات منتشر و دنس در ناحیه ی ساب اپیتلیال به همراه نمای میکروتوبولر یا کریستالی که لوپ های مویرگی را مسدود نموده،مشاهده می شود.
درمان شامل رفع عامل زمینه ای است.مبتلایان به هپاتیت C فعال باید تحت درمان ضد ویروسی قرار گیرند.مبتلایان به گاموپاتی مونوکلونال باید درمان ضدمیلوم دریافت کنند.در کزایوگلوبولینمی Mixed شدید می توان از ریتوکسیماب با یا بدون پلاسما فرز استفاده نمود.
پروگنوز کلیوی خوب بوده و تعداد اندکی به سمت ESRD پیشرفت میکنند.
گلومرولونفریت کرایوگلوبولینمیک:کرایوگلوبولین ها ایمنوگلوبولین هایی هستند که در دمای پایین رسوب می کنند و با گرم شدن دوباره حل می شوند.
اتیولوژی:علل کرایوگلوبولینمی عبارتند از:1- ایدیوپاتیک 2-بیماری های اتوایمیون 3-بدخیمی ها 4- عفونت:عفونت با HVC یکی از علل اصلی کرایوگلوبولینمی است.
این بیماری به سه تایپ بر اساس نوع ایمنوگلوبولین تقسیم میشود.
تظاهرات بیماری:بیماری کلیوی در 20-60% این بیماران رخ می دهد و با پروتئینوری ، هماچوری میکروسکوپیک ،سندرم نفروتیک و اختلال کلیوی تظاهر پیدا میکند.هایپرتنشن شایع بوده و ممکن است به ویژه در صورت همراهی با سندرم نفریتیک حاد،شدید باشد..
از منظر پاتولوژی در میکروسکوپ نوری الگوی ضایعات ممبرانوپرولیفراتیو با واسطه ی کمپلکس ایمنی مشاهده می شود.
در میکروسکوپ الکترونی رسوبات منتشر و دنس در ناحیه ی ساب اپیتلیال به همراه نمای میکروتوبولر یا کریستالی که لوپ های مویرگی را مسدود نموده،مشاهده می شود.
درمان شامل رفع عامل زمینه ای است.مبتلایان به هپاتیت C فعال باید تحت درمان ضد ویروسی قرار گیرند.مبتلایان به گاموپاتی مونوکلونال باید درمان ضدمیلوم دریافت کنند.در کزایوگلوبولینمی Mixed شدید می توان از ریتوکسیماب با یا بدون پلاسما فرز استفاده نمود.
پروگنوز کلیوی خوب بوده و تعداد اندکی به سمت ESRD پیشرفت میکنند.
بیماری کلیه پلی کیستیک اتوزومال غالب
این نوع بیماری در حدود ۹۰ درصد موارد بیماری کلیه پلی کیستیک را شامل می شود. علائم و نشانه های این بیماری اغلب در بین سنین ۳۰ تا ۴۰ سال بروز می دهند به همین دلیل در گذشته، این نوع به بیماری کلیه پلی کیستیک افراد بالغ شناخته می شد.
علائم بیماری:
فشار خون بالا ، درد در کمر یا پهلو ، سردرد ، وجود خون در ادرار ، سنگ های کلیوی ، عفونت های کلیوی یا مجاری ادرار
تشخیص بیماری :
سونوگرافی کلیه ، CT اسکن کلیه ، MRI کلیه
درمان بیماری :
درمان شامل مقابله با علائم، نشانه ها و عوارض این بیماری در سطوح اولیه می شود:
-کنترل فشار خون و رژیم غذایی صحیح
کنترل فشار خون بالا می تواند پیشرفت بیماری را به تاخیر انداخته و روند آسیب کلیوی را کند کند. در صورتی که یک رژیم کم سدیم، کم چربی و دارای مقادیر متوسطی از پروتئین و کالری با عدم مصرف سیگار، افزایش ورزش و کاهش استرس همراه باشد، می تواند به کنترل فشار خون کمک کند.
داروهایی مانند ACEI و ARBI برای کنترل فشار خون موثر می باشند.
-جراحی کلیه پلی کیستیک
-عفونت های کلیه یا مثانه
درمان عفونت ها با دارو های آنتی بیوتیک
-نارسایی کلیوی
انجام دیالیز یا پیوند کلیه نیاز خواهد داشت.
-آنوریسم ها
غربالگری های منظم را برای آنوریسم های درون جمجمه
IgAنفروپاتی
توضیح : نام دیگر آن سندرم برگر است در اطفال شایع تر است ، نوعی سندرم نفریتیک حاد است ، شایع ترین شکل primary GN است و بیماری ایی است که به فیلتر های کوچک داخل کلیه آسیب می زند
علت : وجود نقص در ساختمان IgA است بدین شکل که IgA ساخته شده فاقد گالاکتوز است (IgA1 GD_) بنابراین یک اتوآنتی بادی از جنس IgG و گاهی IgA برعلیه(IgA1 GD_) ساخته می شود و تشکیل کمپلکس ایمنی می دهند و روی سلول های مزانتژیال کلیه رسوب می کنند و به دنبال آن کمپلمان فعال می شود و اگر این روند در کل بدن ایجاد شود سندرم هنوخ شئون لاین ایجاد میشود
علت ثانویه سندروم برگر شامل بیماری مزمن کبدی ، سلیاک ، درماتیت هرپتی فرم ، انکلیوزان اسپوندیلیت
علائم : وجود هماچوری میکروسکوپیک که به صورت تصادفی پیدا می شود و وجود هماچوری gross که اغلب همراه با یک عفونت همزمان مجاری تنفسی ایجاد می شود
وجود پروتئینوری اغلب زیر 500 میلی گرم در روز است
تشخیص : در میکروسکپ نوری تکثیر سلول های مزانژیال و در ایمونوفلورسانس رسوب IgA روی سلول های مزانژیال مشاهده میشود و در میکروسکوپ الکترونی تجمع electron_ dense deposit مشاهده میشود
ریسک فکتور های بد در سندرم برگر : پروتئین اوری بالای 1 گرم ، فشارخون بالا ، اختلال عملکرد کلیه (کراتینین بالا) است
درمان :درموارد خفیف با عملکرد طبیعی کلیه نیاز به درمان ندارد و درمان فقط حمایتی است ولی اگر پروتئین اوری بالای 1 گرم در روز و یا نارسایی پیش رونده کلیه وجود داشته باشد باید از دوز بالای استروئید با یا بدون سیستوتوکسیک استفاده کرد
IgAنفروپاتی
توضیح : نام دیگر آن سندرم برگر است در اطفال شایع تر است ، نوعی سندرم
نفریتیک حاد است ، شایع ترین شکل primary GN است و بیماری ایی است که به
فیلتر های کوچک داخل کلیه آسیب می زند
علت : وجود نقص در ساختمان IgA است بدین شکل که IgA ساخته شده فاقد
گالاکتوز است (IgA1 GD_) بنابراین یک اتوآنتی بادی از جنس IgG و گاهی IgA
برعلیه(IgA1 GD_) ساخته می شود و تشکیل کمپلکس ایمنی می دهند و روی سلول
های مزانتژیال کلیه رسوب می کنند و به دنبال آن کمپلمان فعال می شود و
اگر این روند در کل بدن ایجاد شود سندرم هنوخ شئون لاین ایجاد میشود
علت ثانویه سندروم برگر شامل بیماری مزمن کبدی ، سلیاک ، درماتیت هرپتی
فرم ، انکلیوزان اسپوندیلیت
علائم : وجود هماچوری میکروسکوپیک که به صورت تصادفی پیدا می شود و وجود
هماچوری gross که اغلب همراه با یک عفونت همزمان مجاری تنفسی ایجاد می
شود
وجود پروتئینوری اغلب زیر 500 میلی گرم در روز است
تشخیص : در میکروسکپ نوری تکثیر سلول های مزانژیال و در ایمونوفلورسانس
رسوب IgA روی سلول های مزانژیال مشاهده میشود و در میکروسکوپ الکترونی
تجمع electron_ dense deposit مشاهده میشود
ریسک فکتور های بد در سندرم برگر : پروتئین اوری بالای 1 گرم ، فشارخون
بالا ، اختلال عملکرد کلیه (کراتینین بالا) است
درمان :درموارد خفیف با عملکرد طبیعی کلیه نیاز به درمان ندارد و درمان
فقط حمایتی است ولی اگر پروتئین اوری بالای 1 گرم در روز و یا نارسایی
پیش رونده کلیه وجود داشته باشد باید از دوز بالای استروئید با یا بدون
سیستوتوکسیک استفاده کرد
Stageپاتولوژیک بیماری MPGN:
بیماریMPGN به دو دسته کلی ایدیوپاتیک و ثانویه تقسیم بندی میشود.
MPGN ایدیوپاتیک نیز به سه زیر دسته تقسیم میشود:
Type1\Type2\Type3
(البته یک نوع طبقه بندی اضافی نیز برااساس مکانیسم های پاتولوژیک وجود دارد که به شرح زیر است:
۱_MPGNبا واسطه کمپلکس های ایمنی
۲_MPGNبا واسطه سیستم کمپلمان
۳_MPGNبا مکانیسم هایی که به سیستم کمپلمان یا رسوب ایمونو گلوبولین ها مربوط نمیباشد)
Type1:کمپلکس های ایمنی در فضای ساب اندوتلیال رسوب میکنند و سبب فعال شدن سیستم کمپلکان از مسیر کلاسیک و آلترناتیو و همچنین آزاد سازی انواع کموکاین ها و سایتو کاین ها میشوند.
مدیاتو های التهابی آزاد شده سبب تجمع سلولهای التهابی و پرولیفراسیون سلولهای مزانژیال و اندوتلیال میشوند.این نوع ازMPGN،همچنین با عفونت های ناشی از استافیلوکوک اپیدرمیدیس نیز مرتبط میباشد.
Type2: رسوبات “dense deposite”در غشا پایه گلومرول بوجود می آید.با لیپودیستروفی پارشیال مرتبط است.این نوع از MPGN در اثر رسوب هیچ نوع کمپلکس ایمنی بوجود نمی آید بلکه ازمایشات نوعی رسوبNاستیل گلوکز آمین را نشان می دهند.
رسوب ها همچنین در غشا پایه طحال و غشاbruchشبکیه نیز وجود دارد
Type3:رسوبات در ناحیه ساب اندوتلیال و ساب اپیتلیال بوجود می آیند.رسوب گلومرولارحاویc3-c4و پروپردین است که سبب فعال شدن سیستم کمپلمان از مسیر آلترناتیو میشود،البته فرضیه ای نیز مبنی براینکه تغییرات عروقی درسطه مویرگ ها باعث فعال شدن سیستم کمپلمان می شود وجود دارد.
MCD:شایعترین علت سندرم نفروتیک در کودکان (90-70%)و عامل 20-15 % سندرم نفروتیک در بالغین است .
علت :اغلب ایدیوپاتیک است ندرتا ثانویه به بیماری تیموما وهوچکین یا مصرفNSAID دیده میشود.
علایم بالینی :ادم ،از دست دادن پروتئین محدود به پروتئین های کوچک (عمدتا آلبومین)،فقدان سلول در ادرار و سایر علایم با شیوع کمتر :هایپرتانسیون،هماچوری میکروسکوپی ،آتوپی و نارسایی کلیوی است .
تشخیص:در کودکان بر اساس شک بالینی است در میکروسکوپ نوری به علت تغییرات اندک هیچ یافته ای وجود ندارد توبول ها درجاتی از نفرولیپوز را به صورت سلول های کف الود حاوی چربی نشان میدهند . در میکروسکوپ ایمنوفلوئورسانس چون ایمنوگلوبولین نداریم چیزی نمی بینیم اما در میکروسکوپ الکترونی از بین رفتن زواید پدوسیت دیده میشود .
درمان :اگرچه مشخص شده در 30% کودکان رمیشن خودبه خودی اتفاق می افتد ولی اغلب کودکان با استروئید درمان میشوند.
سیر بیماری:معمولا خود محدود شونده است در کودکان عود پس از اولین رمیشن شایع است و بعد از بلوغ دفعات عود کمتر میشود در کل پیش آگهی در کودکان بهتر از بزرگسالان است.
C1qنفروپاتی یک پروئینوری یا سندرم نفروتیک اسیمپتوماتیک مقاوم به استروئید است.c1qیکی از اجزای سیستم ایمنی است و در این بیماران میتواند در مزانژیوم کلیه رسوب کند یا در ادرار دیده شود و بسیار شبیه بهMCDو FSGSمیباشد.این بیماری بیشتر در بچه ها دیده میشود و شیوع ان در زنان وسیاه پوستان بیشتر است.پاتوفیزلوژی این بیماری مبهم است وعلائم ان شامل:ادم و ورم کردن مخصوصا در کمربند لگنی و HTNو هایپر کلسترولمیا و استعداد به تشکیل لخته ی خون و با یا بدون هماچوری گروس و نارسایی کلیوی.تشخیص بیماری با بیوپسی و در میکروسکوپ نوری بدون ضایعه PGN,FSGSمیباشددر ایمونوفلورسانس C1Qدامیننت یا کودامیننت باFULLHOUSE PATTERN WITH DEPOSITS OF IGG , IGM , IGA,C1Q,C3.درمان ان شامل دارو های کاهنده پروتئین ادرار مثلACEI,ARBSو درمان های سرکوب کننده ایمنی مثل سیکلوسپورین و ازاتیو پرین و تاکرولیموس میباشد.در سیر این بیماری ممکن است به نارسایی کلیه بیانجامد.
IgA نفروپاتی
توضیح : نام دیگر آن سندرم برگر است در اطفال شایع تر است ، نوعی سندرم
نفریتیک حاد است ، شایع ترین شکل primary GN است و بیماری ایی است که به
فیلتر های کوچک داخل کلیه آسیب می زند
علت : وجود نقص در ساختمان IgA است بدین شکل که IgA ساخته شده فاقد
گالاکتوز است (IgA1 GD_) بنابراین یک اتوآنتی بادی از جنس IgG و گاهی IgA
برعلیه(IgA1 GD_) ساخته می شود و تشکیل کمپلکس ایمنی می دهند و روی سلول
های مزانتژیال کلیه رسوب می کنند و به دنبال آن کمپلمان فعال می شود و
اگر این روند در کل بدن ایجاد شود سندرم هنوخ شئون لاین ایجاد میشود
علت ثانویه سندروم برگر شامل بیماری مزمن کبدی ، سلیاک ، درماتیت هرپتی
فرم ، انکلیوزان اسپوندیلیت
علائم : وجود هماچوری میکروسکوپیک که به صورت تصادفی پیدا می شود و وجود
هماچوری gross که اغلب همراه با یک عفونت همزمان مجاری تنفسی ایجاد می
شود
وجود پروتئینوری اغلب زیر 500 میلی گرم در روز است
تشخیص : در میکروسکپ نوری تکثیر سلول های مزانژیال و در ایمونوفلورسانس
رسوب IgA روی سلول های مزانژیال مشاهده میشود و در میکروسکوپ الکترونی
تجمع electron_ dense deposit مشاهده میشود
ریسک فکتور های بد در سندرم برگر : پروتئین اوری بالای 1 گرم ، فشارخون
بالا ، اختلال عملکرد کلیه (کراتینین بالا) است
درمان :درموارد خفیف با عملکرد طبیعی کلیه نیاز به درمان ندارد و درمان
فقط حمایتی است ولی اگر پروتئین اوری بالای 1 گرم در روز و یا نارسایی
پیش رونده کلیه وجود داشته باشد باید از دوز بالای استروئید با یا بدون
سیستوتوکسیک استفاده کرد
بیماری کلیه پلی کیستیک اتوزومال مغلوب(نوع کودکان):
ARPKD یک اختلال نادر اتوزوم مغلوب است که از نظر ژنتیکی از بیماری بالغین متمایز است.
تمام انواع این بیماری،از جهش در ژن PKHD1 که یک پروتئین گیرنده غشایی معروف به فیبروسیستین( Fibrocystin) را کد میکند منشا میگیرند.
*نمای پاتولوژیک: کلیه دارای کیست های متعدد کوچک در کورتکس و مدولا بوده که نمای اسفنجی به کلیه میدهد. کانال های متسع و کشیده شده به صورت عمود بر سطح کورتکس وجود دارندکه به صورت کامل جانشین بافتهای مدولا و کورتکس میشوند. کیست ها دارای پوشش یک شکل سلول های مکعبی هستند. بیماری همیشه دوطرفه است.
*ویژگی های بالینی: ARPKD کلاسیک به طور کامل در داخل رحم یا طی نوزادی تشخیص داده میشود و مشخصه آن وجود کلیه های به شدت بزرگ اکوژن در جنین های مبتلا است. کاهش تولید ادرار در جنین میتواند سبب وقوع اولیگوهیدر آمنیوس و هایپوپلازی ریه شود.
برخی بیماران پس از طی مرحله نوزادی تشخیص داده میشوند و گروه سنی بالاتری دارند. عوارض این گروه اغلب شامل فشار خون بالای سیستمیک، نارسایی پیشرونده کلیه، و تظاهرات کبدی است.شاه علامت های بیماری کبدی ARPKD عبارتاند از دیس ژنزی صفراوی همراه با فیبروز پریپورتال مرتبط با آن، که فیبروز کبدی مادرزادی(CHF) نامیده میشود، و اتساع مجاری صفراوی داخل کبدی(بیماری کارولی)
*تشخیص: برای تشخیص میتوان از سونوگرافی، CT، و MRI ، استفاده کرد. در سونوگرافی کلیه های بزرگ و اکوژن مشاهده میشوند که تمایز قشر- مدولا در آنها ضعیف است. در موارد شدید پس از هفته ۲۴جنینی میتوان تشخیص را در داخل رحم مطرح کرد.
*درمان: درمانی اختصاصی برای ARPKD وجود ندارد. مراقبت های ویژه ی مناسب در دوران نوزادی ، کنترل فشار خون ، دیالیز، و پیوند کلیه میزان بقارا تا بزرگسالی افزایش داده اند. عوارض فیبروز کبدی ممکن است انجام پیوند کبد را ضروری سازند. بیماران مبتلا به بیماری شدید کارولی ممکن است نیاز به شانت پورتوسیستمیک داشته باشند.
IgAنفروپاتی
توضیح : نام دیگر آن سندرم برگر است در اطفال شایع تر است ، نوعی سندرم نفریتیک حاد است ، شایع ترین شکل primary GN است و بیماری ایی است که به فیلتر های کوچک داخل کلیه آسیب می زند
علت : وجود نقص در ساختمان igA است بدین شکل که igA ساخته شده فاقد گالاکتوز است (_GD _IgA1) بنابراین یک اتوآنتی بادی از جنس igG و گاهی igA برعلیه(IgA1 GD_) ساخته می شود و تشکیل کمپلکس ایمنی می دهند و روی سلول های مزانتژیال کلیه رسوب می کنند و به دنبال آن کمپلمان فعال می شود و اگر این روند در کل بدن ایجاد شود سندرم هنوخ شئون لاین ایجاد میشود
علت ثانویه سندرم برگر شامل بیماری مزمن کبدی ، سلیاک ، درماتیت هرپتی فرم ، انکلیوزان اسپوندیلیت
علائم : وجود هماچوری میکروسکوپیک که به صورت تصادفی پیدا می شود و وجود هماچوری gross که اغلب همراه با یک عفونت همزمان مجاری تنفسی ایجاد می شودوجود پروتئینوری اغلب زیر 500 میلی گرم در روز است
تشخیص : در میکروسکپ نوری تکثیر سلول های مزانژیال و در ایمونوفلورسانس رسوب igA روی سلول های مزانژیال مشاهده میشود و در میکروسکوپ الکترونی تجمع electron_ dense deposit مشاهده میشود
ریسک فکتور های بد در سندرم برگر : پروتئین اوری بالای 1 گرم ، فشارخون بالا ، اختلال عملکرد کلیه (کراتینین بالا) است
درمان :درموارد خفیف با عملکرد طبیعی کلیه نیاز به درمان ندارد و درمان فقط حمایتی است ولی اگر پروتئین اوری بالای 1 گرم در روز و یا نارسایی پیش رونده کلیه وجود داشته باشد باید از دوز بالای استروئید با یا بدون سیستوتوکسیک استفاده کرد
گلومرولواسکلروز فوکال سگمنتال (FSGS) :
از نظر بافت شناسی با اسکلروز برخی گلومرول ها و نه تمام انها و ابتلای فقط قسمت هایی از هر گلومرول مشخص میشود.
عامل ۱۰ تا ۱۵ درصد موارد سندرم نفروتیک کودکان و. ۲۵ درصد موارد در بالغین است.
اتیولوژی: ۱-اولیه یا ایدیوپاتیک (شایع ترین) ۲-ثانویه به عللی مانند ایدز، ریفلاکس ادراری ، کم خونی، سیل سل و … ۳- خانوادگی ۴- داروها مانند اینترفرون ، پامیدرونات ، سیرولموس
پاتولوژی: بیشتر گلومرول های واقع در محل کورتکس و مدولا درگیر میشوند.
پیش اگهی و درمان: رمیشن خودبخودی در FSGS نادرست است و پاسخ به استروئید تنها در ۲۰ تا ۴۰ درصد موارد دیده میشوند.
در موارد ثانویه علل زمینه ای باید درمان شوند.
در موارد اولیه استروئید سیستمیک به مدت ۶ تا ۹ ماه و مهارکننده های رنین- انژیوتانسین توصیه میشود.
اتیولوژی: ۱-اولیه یا ایدیوپاتیک (شایع ترین) ۲-ثانویه به عللی مانند ایدز، ریفلاکس ادراری ، کم خونی، سیل سل و … ۳- خانوادگی ۴- داروها مانند اینترفرون ، پامیدرونات ، سیرولموس
پاتولوژی: بیشتر گلومرول های واقع در محل کورتکس و مدولا درگیر میشوند.
پیش اگهی و درمان: رمیشن خودبخودی در FSGS نادرست است و پاسخ به استروئید تنها در ۲۰ تا ۴۰ درصد موارد دیده میشوند.
در موارد ثانویه علل زمینه ای باید درمان شوند.
در موارد اولیه استروئید سیستمیک به مدت ۶ تا ۹ ماه و مهارکننده های رنین- انژیوتانسین توصیه میشود.
سندرم نفروتیک مادرزادی نوع اسکلروز مزانژیال منتشر (DMS) یکی از دلایل ایجاد سندرم نفروتیک در دوره کودکی و نوزادی می باشد که علائم آن در بررسی پاتولوژی عبارتند از: اسکلروز پیشرونده ی ماتریکس مزانژیال، هایپرتروفی پودوسیت ها در بیماری اولیه، ضخیم شدن غشای پایه، کاهش قطر لومن مویرگ و ظاهر دندانی شکل پودوسیت ها در بیماری پیشرفته.
فرم های ژنتیکی نفروپاتی مانند DMS معمولا به درمان های ایمونوساپرسیو مقاوم اند و به سرعت به سمت ESKD پیشرفت می کنند.
جهش ژنتیکی در ژن های WT1 و PLCE1 در DMS یافت شده اند.
علائم DMS همانند سایر انواع سندرم نفروتیک شامل پروتئینوری، هیپوآلبومینمی، ادم و هیپرلیپیدمی می باشد که در سه ماهه اول زندگی بروز می یابد.
علت سندرم نفروتیک مادرزادی ممکن است عفونت های داخل رحمی یا تجویز برخی داروها طی بارداری باشد.
این اختلال ممکن است قبل از تولد به وسیله بالارفتن سطح آلفافیتوپروتئین در خون مادر یا مایع آمنیوتیک تشخیص داده شود.
پلی آنژئیت میکروسکوپی :
پلی آنژئیت میکروسکوپی نوعی واسکولیت است که با التهاب عروقی و علایم متعددی همراه است .
میانگین سن آغاز بیماری تقریبا 57 سال است و مردان با فراوانی اندکی بیش تر از زنان مبتلا می شوند .
این بیماری تمایل به درگیر کردن مویرگ ها و وریدچه ها به همراه شریان ها با اندازه کوچک و متوسط دارد .
رنگ آمیزی ایمونوهیستوشیمیایی نشانگر اندک بودن رسوب ایمونوگلوبین ها در ضایعه ی عروقی پلی آنژئیت میکروسکوپی است ، که دلالت بر آن دارد که تشکیل کمپلکس ایمنی نقشی در این سندرم ندارد .
ضایعه ی کلیوی موجود در این واسکولیت همان است که در وگنر یافت می شود . این بیماری به شدت با حضور P-ANCA ارتباط دارد .
شروع بیماری می تواند تدریجی همراه با علائم ابتدایی نظیر تب ، کاهش وزن و درد عضلانی – اسکلتی و آرترالژی همراه باشد . با این حال شروع بیماری اغلب حاد است .
گلومرونفریت در دست کم 79 درصد از بیماران روی می دهد ومی تواند به سرعت پیش رونده باشد. که در نهایت به نارسایی کلیوی ختم می گردد .
خلط خونی می تواند نخستین علامت خون ریزی آلوئولی ریه باشد که در 12 درصد از بیماران روی می دهد .سایر علائم این بیماری شامل منونوریت مولتی پلکس و بیماری گوارشی و بیماری پوستی هستند .بیماری راه های تنفسی فوقانی و ندول های ریوی معمولا در این واسکولیت یافت نمی شوند و اگر وجود داشته باشند بر بیماری وگنر دلالت دارند .
در بیماری پلی آنژئیت میکروسکوپی علائم و نشانه های التهاب ممکن است دیده شوند . که شامل ESR ، کم خونی یا آنمی ، لکوسیتوز و ترومبوسیتوز هستند .
تشخیص این بیماری بر اساس شواهد بافت شناسی واسکولیت یا گلومورونفریت pauci-immune در یک بیمار با علایم واسکولیت می باشد .
بیماران مبتلا به بیماری شدید و کشنده باید با ترکیب پردنیزولون و سیکلوفسفامید یا ریتوکسیماب روزانه تحت درمان قرار بگیرند
تومور ویلمز (پروژه جلسه دوم)
اگرچه تومور ویلمز در بالغین نادر است، ولی سومین سرطان شایع توپور (غیرهماتولوژیک) در کودکان کمتر در ۱۰ سال است. این تومورها حاوی انواعی از اجزای سلولی و بافتی هستند که همگی از مزودرم مشتق شده اند. تومور ویلمز مثل رتینوبلاستوم می تواند خانوادگی یا تک درگیر باشد و استعداد تومورزایی می تواند به صورت صفت اتوزومی غالب منتقل شود. این تومور از بلاستما نفروژنیک حاضر در کلیه در حال تکامل منشا می گیرد. تومور ویلمز از نظر پاتولوژی از سه جز بافت تشکیل شده است؛ جزء بلاستما، جزء اپیتلیال و جزء استرومال؛ گرچه تومور می تواند بای فازیک یا در شرایط نادر مونوفازیک باشد. بخش بلاستما که از سلول های تمایز نیافته بیضی شکل با هسته های هایپرکروماتیک تشکیل شده، می تواند نمای ندولار، پیچ در پیچ یا منتشر داشته باشد. بلاستما می تواند به صورت محدود یا غالب در تومور موجود باشد. بخش اپیتلیال می تواند اشکال گوناگونی داشته باشد، اما اکثراً توبول های ناشی از آرایش سلول های استوانه ای بلند اولیه تشکیل شده است که ظاهری شبیه گل لباس دارد. اپیتلیوم مژک دار و متاپلازی سنگفرشی هم ممکن است دیده شود. بخش استرومال از فردی به فرد دیگر نمای متفاوتی دارد، اما می تواند از اجزای هتروژن مثل غضروف، استخوان و عضله اسکلتی تشکیل شده باشد. اکثر تومورها به صورت تک گیر بروز می کنند، ولی حدود ۱۰ درصد آن ها با اختلالات مادرزادی خاص مثل سندروم WARG، سندروم Denys-Drash و سندروم Beckwith-Wiedemann ارتباط دارد. / منبع: pathpedia.com
نوروبلاستوما
یکی از شایعترین تومورهای بدخیم توپر دوران کودکی است که در کلیه ایجاد شده و بیشترین احتمال بروز آن در کودکان وجود دارد. منشأ این تومور نورواندوکرین از سلولهای ستیغ عصبی (اجزای سیستم سمپاتیک) است. معمولاً نوروبلاستوما در کودکان کمتر از 5 ساله تشخیص داده می شود و در بیشتر موارد کودک مبتلا کمتر از یک سال دارد. شایعترین علامت نوروبلاستوما، وجود یک توده یا برآمدگی غیرمعمول است که معمولاً در شکم کودک یافت میشود. سایر علائم عبارتند از تب، اسهال مقاوم، فشارخون بالا (به دلیل تحریک پذیری)، ضربان سریع قلب، سرخی پوست، گر گرفتگی و تعریق. گاه چشمان کودک به شکلی غیرطبیعی حرکت میکنند. این وضعیت با نام نشانگان اپسوکلونوس – میوکلونوس شناخته میشود. تشخیص نوروبلاستوما: در نود درصد موارد نوروبلاستوما، سلولهای تومور کاتکول آمینهاها را به حدی ترشح میکنند که در ادرار و خون قابل سنجش باشند. بعضی علایم مرتبط با نوروبلاستوما هم چون فشار خون بالا، ضربان سریع قلب یا اسهال، مستقیماً به دلیل افزایش کاتکول آمینها میباشند. تشخیص قطعی نیازمند دیدن سلولهای نوروبلاستوما در نمونههای بافتی زیر میکروسکوپ است. به این منظور بیوپسی مغز استخوان یا آسپیراسیون آن انجام میگیرد. بهطور کلی داروها در دو سوم کودکان کاملاً جواب خواهند داد. ترکیبی که امروز به کار میرود عبارت است از: سیکلوفسفامید به علاوه دوکسوروبیسین یا سیس پلاتین به علاوه اتوپوساید..
نفروبلاستوماتوز (پروژه جلسه دوم)
نفروبلاستوماز nephrogenic rests (بخشی از بافت جنینی که بعد از تکامل کلیه در آن باقی می ماند) منتشر یا چند کانونه هستند که در حدود ۱ درصد اطفال گزارش شده است. nephrogenic rests کانون هایی از بلاستوماهای متانفریک هستند که در ۳۰ تا ۴۰ موارد به تومور ویلمز تبدیل می شوند. در بررسی gross ممکن است تظاهر خاصی نباشند یا ندول های حاصل از ضخیم شدگی کپسول یا تشکیل اسکار در زیر کپسول، کورتکس و مدولا مشخص شود. در بررسی میکروسکوپی، کلنی های دسته ای یا منتشر از سلول های اولیه (primitive) دیده می شود. غضروف یا مزانشیم اولیه مشاهده نمی شود. ممکن است الگوی اسکلروز یا هایپرپلاستیک داشته باشد. در نوع بین لوبی، سلول ها به صورت پراکنده در کورتکس و مدولا پخش شده اند و حاشیه های نامنظم دارند. میزان استروما نیز نسبت به بلاستوما و توبول ها بیشتر است. در نوع اطراف لوبولی، سلول ها در محیط با مرزهای مشخص قرار دارند و از بلاستوما و توبول ها با استرومای کم یا اسکلروتیک تشکیل شده اند.
مولتی لوکولار کیستیک نفروما (پروژه جلسه دوم)
( multiloculated cystic nephroma )
شامل کیست های متعدد می باشد خوش خیم است،
توزیع سنی دوگانه دارد.در سن ۲-۳سال و دهه ۵-۶ زندگی شایع است.
در بچه ها در جنس مذکر و در سن بالا در زنان شایع است.
در بچه ها بدون علامت یا به شکل توده شکم و در بالغین به شکل درد،هماچوری و عفونت ادراری مشخص می شود.
در رادیولوژی به شکل کیست های مولتی لوکاله و متعدد هستندکه در تقسیم بندی بوسنیاک در کلاس III و مشکوک به بدخیمی هستند .اغلب این ضایعات با نفرکتومی رادیال یا پارشیال درمان می شوند.
آمیلوئیدوز کلیوی(قسمت اول)
یا در نتیجه ی رسوب های فیبریلار زنجیره ی سبک ایمونوگلوبولین (آمیلوئیدوز اولیه) یا رسوب های فیبریلار قطعات پروتئین آمیلوئید A (آمیلوئیدوز ثانویه). گرچه هر دو به دلایل متفاوتی رخ می دهند پاتوفیزیولوژی بالینی کاملا مشابه است.
در نوع اولیه زنجیره های سبک که در دیسکرازی پلاسماسل به مقدار زیاد تولید شده اند توسط ماکروفاژ ها به قطعاتی تقسیم شده اند که در PH اسیدی می توانند جمع شوند و بیشتر از نوع لامبدا هستند(75 درصد) حدود 10 درصد از این بیماران میلوم آشکار همراه ضایعات سیستیک استخوانی و ترشح مغز استخوان با بیش از 30 درصد پلاسماسل دارند در این حالت سندرم نفروتیک شایع است و حدود 20 درصد بیماران به سمت دیالیز می روند.
آمیلوئیدوز ثانویه می تواند با سندرم نفروتیک تظاهر کند که ناشی از رسوب صفحات چین خورده ی B پروتئین آمیلوئید A سرم است که یک واکنشگر فاز حاد است و عملکرد فیزیولوژیک آن شامل انتقال کلسترول جذب سلول ایمنی و فعال کردن متالوپروتئیناز ها است.40 درصد بیماران آمیلوئید AA آرتریت روماتویید دارند 10 درصد دیگر اسپوندیلیت انکیلوزان و آرتریت پسوریاتیک دارند و بقیه ناشی از دلایل کمتر شایع دیگر است همچنین در نوع ثانویه بیماری التهابی روده ، عفونت مزمن یا تب مدیترانه ای فامیلی بسیار شایع است.
آمیلوئیدوز کلیوی (قسمت دوم)
شایعترین رویکرد تعیین نوع آمیلوئید شامل ایمونوفلوروسانس یا ایمونوهیستوشیمی است اما آزمایش ژنتیکی و اسپکترومتری توده کروماتوگرافی مایع نیز برای تعیین نوع آمیلوئید با وضوح بالا مفید است.
درمان: بستگی به منشا پروتئین آمیلوئید دارد در بیماران آمیلوئید AL درمان ضد میلوما با دوز بالای ملفالان و پیوند سلول بنیادی اتولوگ مفید است در موارد انتخابی منجر به دفع بیماری می شود در آمیلوئیدوز ثانویه در فرایند التهاب زمینه ای با داروهای ضد میکروبی یا ضد التهابی انجام می شود.
تومور رابدوييد (RT) یک سارکوم بافت نرم کودکان است که در کلیه ، کبد ، اعصاب محیطی و کلیه قسمتهای نرم دیگر در بدن ایجاد می شود. RT اي که شامل سیستم عصبی مرکزی (CNS) است ، تومور atypical teratoid rhabdoid نامیده می شود. RT معمولاً در دوران كودكی (زير2 سال) رخ می دهد. 90٪ موارد RT با غیرفعال شدن ژن SMARCB1 (22q11.23) كه ژن سرکوبگر تومور است مرتبط است.
هنگامی که تومور با اسکنهای تصویربرداری مشخص شد با بيوپسي تشخيص را تاييد مي كنيم. در بيوپسي تومور تکثیر پراکنده سلولهای گرد یا چند ضلعی با هسته های خارج از مرکز ، هسته های برجسته و سیتوپلاسم ائوزینوفیلی شیشه ای حاوی اجسام هیالین تشکیل شده است که در ورقه ها قرار گرفته اند ديده مي شود.
هیچ درمان استاندارد برای RT وجود ندارد درمان شامل برداشتن توده تومور (تا حد امکان کامل) ، شیمی درمانی تها جمي و هر وقت ممکن باشد رادیوتراپی است.
میزان بقاي 5 ساله ي بيماران زير بيست درصد است و عوامل پیش آگهی شامل متاستاز ، سن كم در تشخیص (<2 سال) و برداشتن ناقص است.
نحوه ی اپروچ به کیست های کلیوی:
تشخیص به طور تیپیک با مشاهده وجود سابقه خانوادگی مثبت مطابق با توارث اتوزومال غالب و وجود کیست های متعدد در
هر دو کلیه مطرح می شود.جهت تشخیص و مشاهده کیست ها اغلب سونوگرافی انجام می شود. اما با این حال حساسیت CT-SCAN و MRI بر پایه T2 با یا بدون ماده حاجب بیش از سونوگرافی است و اطلاعات دقیق تری از مورفولوژی کیست در
اختیار ما قرار می دهد . و می تواند کیست هایی با اندازه کوچک تر را شناسایی کند
طبقه بندی بوسنیاک جهت بررسی و اپروچ به کیست های کلیوی به طور خلاصه :
بوسنیاک I همان کیست ساده و خوش خیم می باشد و نیاز به اقدام خاصی ندارند .
بوسنیاک II ¬ این گروه هم خوش خیم هستند ولی نیاز به پیگیری دارند .
بوسنیاک III-IV آن گروه از کیست ها که در این دو دسته قرار می گیرند با توجه به احتمال بالای بدخیمی در اینها
توصیه به بررسی دقیق و حتی گاهی نیازمند جراحی و خروج تمام و یا قسمتی از کلیه می باشند
Iga nephropathy
Glomerolonephrit cresentic
(RpGN)
چند نوع دارد
شامل علل اوليه يا ايديوپاتيك و علل ثانويه .
علل اوليه شامل:
١.antiglumerular basement membrane
كه با خطوط رسوبي(linear) از ايمونوگلوبين G مشخص ميشود .
٢.Immune complexmediator كه با رسوبات گرانولار از ايمونوگلوبين ها مشخص ميشود.
٣.pauci immune
با رسوبات antineutrophil cytoplasmic antibody همراه با واسكوليت رگ هاي كوچك كه ميتواند ثانويه به يك بيماري سيستميك مثل وگنر باشد.
٤.تركيبي از نوع ١و٣است.
٥.ANCA negative pauci immune renal vasculitis
كرسنتريك هلالي از پروليفراسيون سلول هاي اپيتليال و انفيلتراسيون ماكروفاژها كه كپسول بومن را ميپوشانند و نشاندهنده آسيب گلومرولي شديد است.
نوع ثانويه RPGN ممكن است با عفونت هايي مثل هپاتيت B،استرپتوك،عفونت اندوكارديت،داروها يا ثانويه به بيماري هاي گلومرولي مثل ميلوپروليفراتيو است.
ادامه آمیلوئیدوز :
تب مدیترانهای فامیلی (fmf) به صورت غیرشایع در کشورهای غربی ولی با شیوع بیشتر در نواحی مدیترانهای بخصوص یهودیان سفاردیک و عراقی دیده میشود . fmf به علت موتاسیون در ژن کدکننده پیرین بوجود میآید ، در حالی که سندرم ماکل-ول یک اختلال مشابه از موتاسیون در کرایوپیرین است ، هردو پروتئین در آپوپتوز اولیه لوکوسیتها در التهاب مهم هستند . چنین پروتئینهایی با زنجیره پیرین بخشی از یک مسیر جدیدند که inflammasome نامیده میشوند . موتاسیون رسپتور در سندرم TNFR1- دورهای نیز التهاب مزمن و التهاب ثانویه بوجود میآورد .
بیوپسی از کلیه یا کبد درگیر در 90% موارد که احتمال پیش از تست بالاست تشخیصی است ، آسپیراسیون توده چربی شکمی در 70% موارد مثبت است ، اما ظاهرا زمانی که بدنبال آمیلوئید AA باشیم ، کمتر میباشد . رسوبهای آمیلوئید در طول عروق خونی و نواحی مزانژیال کلیه پخش شدهاند .
نفروم مزوبلاستیک
شایع ترین تومور کلیوی کودکان در دوران نوزادی.
5٪ کل تومورهای کلیوی کودکان و به ندرت در کودکان بزرگتر از 2 سال رخ می دهد.
سه نوع دارد:
كلاسيك:تومور توبولها و گلومرولها را احاطه کرده است ، دارای مرزهای نامنظم است. متاپلازی کندروپلازی / دیسپلازی متداول است. میتوز نادر است؛ نکروز / دسموپلازی وجود ندارد.
سلولی:تکثیر ورق مانند گلبول ، سلولهای دوقلوی آتیپیک با سیتوپلاسم فراوان ، هسته های وزیکولار و نوکلئولها است. ارقام میتوزیک مکرر و نکروز. تومور مرز تحرک دارد.
مختلط: تومورها با ترکیبی از ویژگیهای فوق.
درمان:نفرکتومی با حاشیه های گسترده_ شیمی درمانی در صورتی که برداشتن در نوزادان 3 ماه یا بالاتر ناقص باشد ، یا اگر پارگی تومور در حین عمل شود.
کارسینوما سلول کلیوی روشن (CCRCC) یک تومور قشر کلیوی است که معمولاً توسط سلولهای اپیتلیال بدخیم با سیتوپلاسم واضح و الگوی رشد جمع و جور (آشیانه) یا آکینار (تو در تو در تو در تو در تو) همراه با عروق پیچیده و آربورایز مشخص می شود. تعداد متغیر سلولهای دارای سیتوپلاسم ائوزینوفیلیک دانه ای ممکن است وجود داشته باشد.
ارائه ناخالص معمولی کارسینوما سلول کلیوی روشن (پایین سمت چپ) با رنگ طلایی به دلیل تجمع لیپید داخل سلولی. مناطق سفید کانونی از تمایز سارکوماتوئید هستند. به تومور در ورید کلیه توجه داشته باشید.